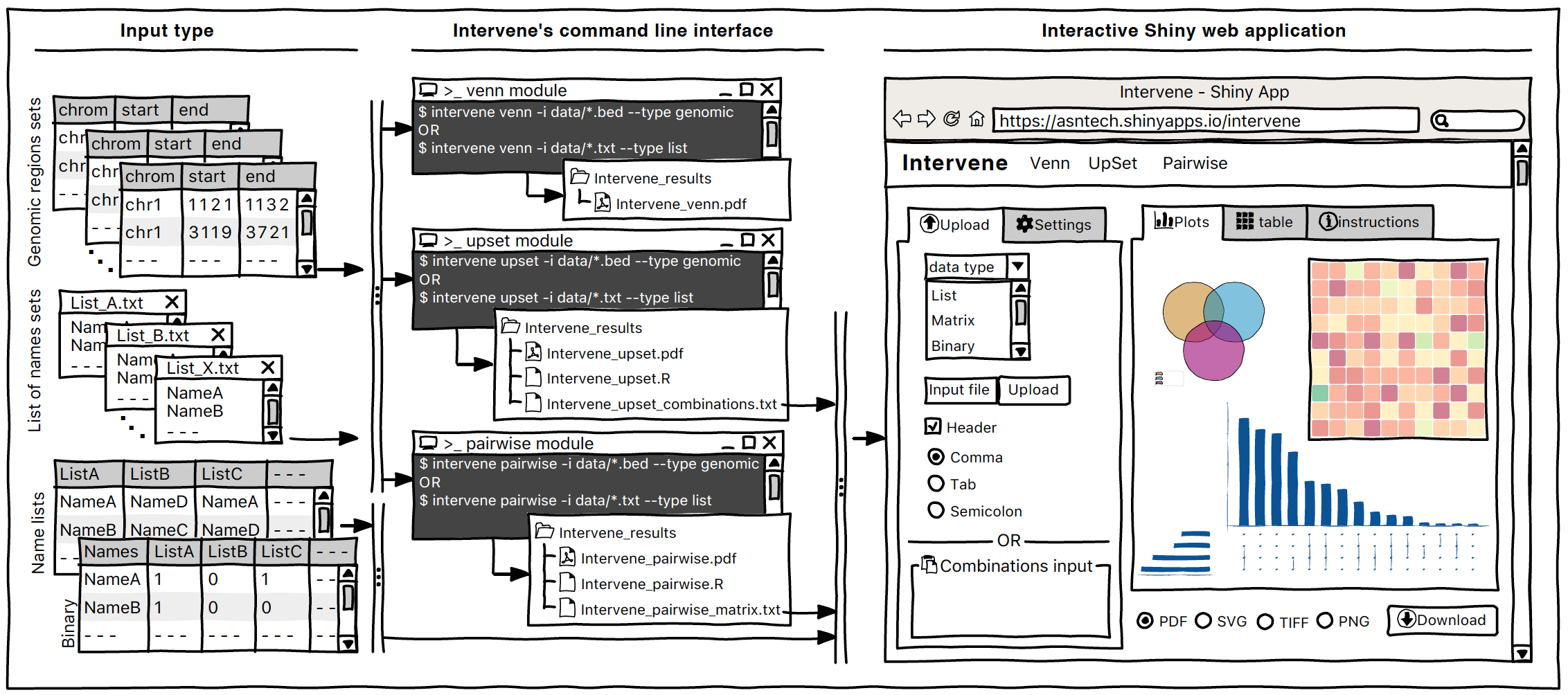
a tool for intersection and visualization of multiple gene or genomic region sets






A detailed documentation is available in different formats: HTML | PDF | ePUB
conda install -c bioconda interveneThis will install all the dependencies and you are ready to use Intervene.
You can install Intervene from PyPi using pip.
Install from PyPi:
pip install intervene
Note: If you install using pip, make sure to install BEDTools and R packages listed below.
Intervene requires the following Python modules and R packages:
- Python (=> 3.3 ): https://www.python.org/
- BedTools (Latest version): https://github.com/arq5x/bedtools2
- pybedtools (>= 0.7.9): https://daler.github.io/pybedtools/
- Pandas (>= 0.16.0): http://pandas.pydata.org/
- Seaborn (>= 0.7.1): http://seaborn.pydata.org/
- R (>= 3.0): https://www.r-project.org/
- R packages including UpSetR (v1.4.0), corrplot
We are using pybedtools, which is Python wrapper for BEDTools. So, BEDTools should be installed before using Intervene. It's recomended to have a latest version, but if you have an older version already install, it should be fine.
A quick installation, if you have conda installed.
conda install -c bioconda bedtoolsPlease read the instructions at https://github.com/arq5x/bedtools2 to install BEDTools, and make sure it is on your path and you are able to call bedtools from any directory.
Intervene rquires three R packages, UpSetR , corrplot for visualization and Cairo to generate high-quality vector and bitmap figures.
install.packages(c("UpSetR", "corrplot", "Cairo"))You can install a development version by using git from GitHub or Bitbucket.
If you have git installed, use this:
git clone https://bitbucket.org/CBGR/intervene.git
cd intervene
python setup.py sdist installIf you have git installed, use this:
git clone https://github.com/asntech/intervene.git
cd intervene
python setup.py sdist installOnce you have installed Intervene, you can type:
intervene --helpThis will show the following help message.
usage: intervene <subcommand> [options]
positional arguments <subcommand>:
{venn,upset,pairwise}
List of subcommands
venn Venn diagram of intersection of genomic regions or list sets (upto 6-way).
upset UpSet diagram of intersection of genomic regions or list sets.
pairwise Pairwise intersection and heatmap of N genomic region sets in <BED/GTF/GFF> format.
optional arguments:
-h, --help show this help message and exit
-v, --version show program's version number and exitto see the help for the three subcommands pairwise, venn and upset type:
intervene pairwise --help
intervene venn --help
intervene upset --helpTo run Intervene using example data, use the following commands. To access the test data make sure you have sudo or root access.
intervene pairwise --test
intervene venn --test
intervene upset --testIf you have installed Intervene locally from the source code, you may have problem to find test data. You can download the test data here https://github.com/asntech/intervene/tree/master/intervene/example_data and point to it using -i instead of --test.
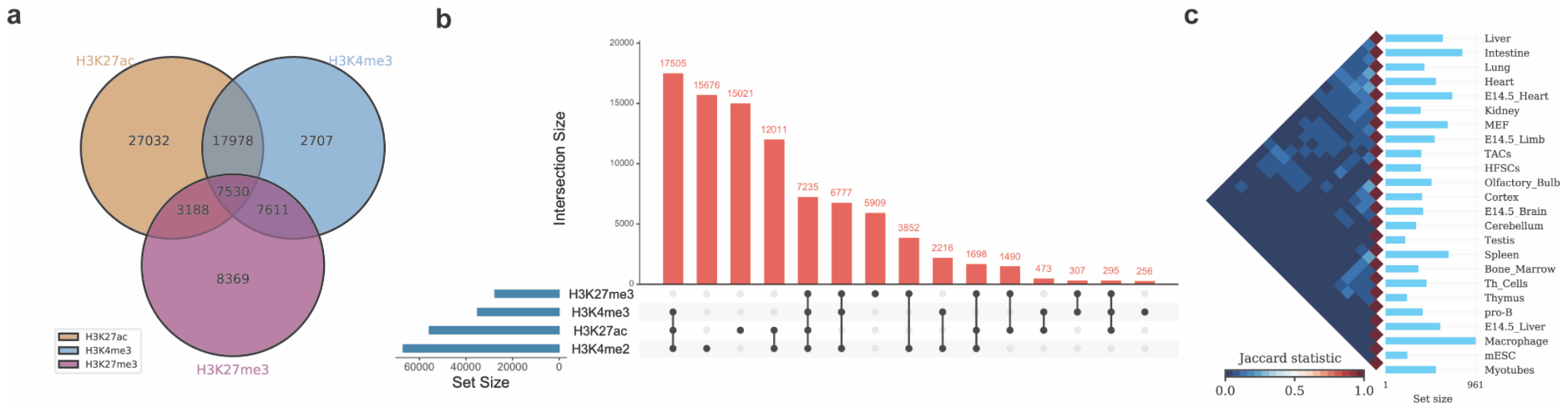
./intervene/intervene venn -i intervene/example_data/ENCODE_hESC/*.bed
./intervene/intervene upset -i intervene/example_data/ENCODE_hESC/*.bed
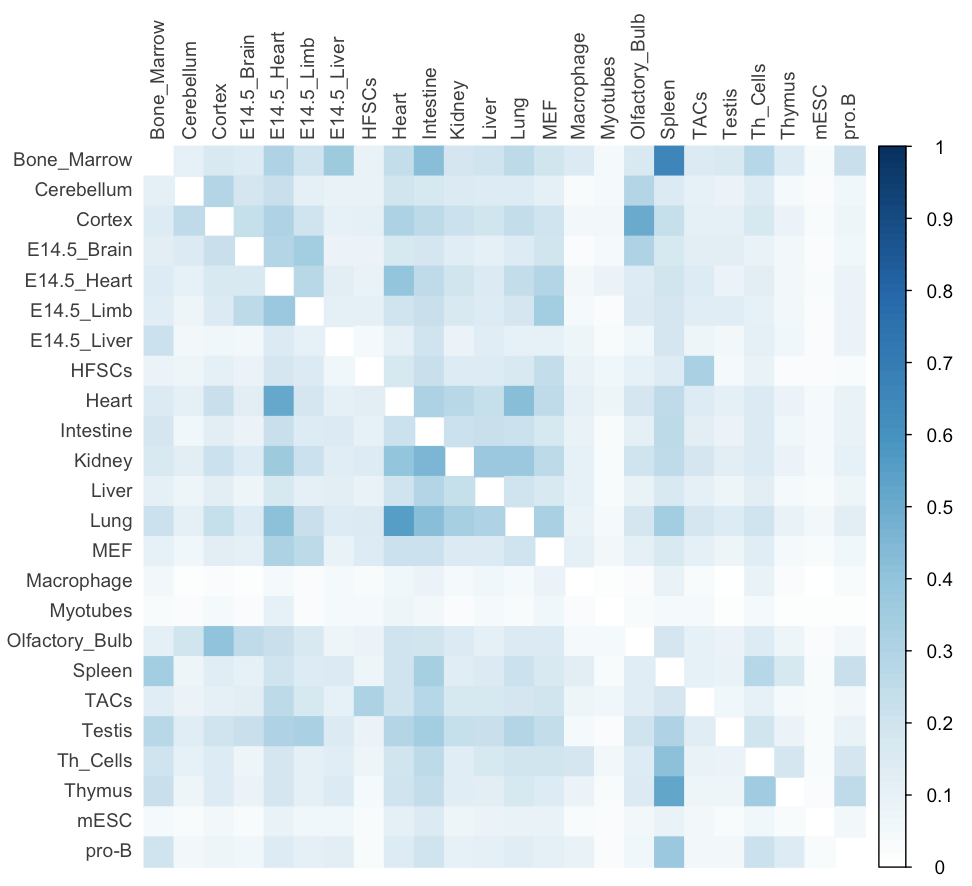
./intervene/intervene pairwise -i intervene/example_data/dbSUPER_mm9/*.bedThe above three test commands will generate the following three figures (a, b and c).
By default your results will stored in the current working directory with a folder named Intervene_results. If you wish to save the results in a specific folder, you can type:
intervene upset --test --output ~/path/to/your/folder
Intervene Shiny App is freely available at https://asntech.shinyapps.io/intervene or https://intervene.shinyapps.io/intervene
The source code for the Shiny app is available at https://github.com/asntech/intervene-shiny
If you have questions, or found any bug in the program, please write to us at azez.khan[at]gmail.com
If you use Intervene please cite us: Khan A, Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinformatics. 2017;18:287. doi: 10.1186/s12859-017-1708-7

















































































































{kind=link}