haowenz / chromap Goto Github PK
View Code? Open in Web Editor NEWFast alignment and preprocessing of chromatin profiles
Home Page: https://haowenz.github.io/chromap/
License: MIT License
Fast alignment and preprocessing of chromatin profiles
Home Page: https://haowenz.github.io/chromap/
License: MIT License










































































































I am trying to map chipseq data, without providing a barcode file. chromap fails after mapping with the following error:
Numbers of reads and barcodes don't match!
Does chromap only work with barcoded chipseq data? Otherwise what might I be doing wrong?
This is the command I am using:
chromap --skip-barcode-check -t 64 --preset chip -x ../atacseq/index -r hg19.fa -1 <fastq.gz> -2 <fastq.gz>
-o <bed>
Many thanks!
Using the pre-compiled version I get the following error:
./chromap --preset hic -x test.index -r ref.fa -1 r1.fq -2 r2.fq -o out.sam --SAM
Preset parameters for Hi-C are used.
Start to map reads.
Parameters: error threshold: 4, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 1000, MAPQ-threshold: 1, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 1
Analyze bulk data.
Won't try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Allow split alignment.
Output mappings in SAM format.
Reference file: ref.fa
Index file: test.index
1th read 1 file: r1.fq
1th read 2 file: r2.fq
Output file: out.sam
Loaded all sequences successfully in 4.05s, number of sequences: 1924, number of bases: 1679214157.
Kmer size: 17, window size: 7.
Lookup table size: 177152077, occurrence table size: 309193718.
Loaded index successfully in 7.85s.
Mapped all reads in 0.53s.
Number of reads: 0.
Number of mapped reads: 0.
Number of uniquely mapped reads: 0.
Number of reads have multi-mappings: 0.
Number of candidates: 0.
Number of mappings: 0.
Number of uni-mappings: 0.
Number of multi-mappings: 0.
Segmentation fault
The fastq files are not empty as the error message would suggest, nonetheless they are non-canonical HiC but 4C, actually, and they are derived from a rather unbalanced read length between mates, r1=28 vs r2=120. Could this be violating some assumptions of the software?
head r1.fq
@NS500648:585:HK55NAFX2:1:11101:9001:1025 1:N:0:ACAAGGTA
GCCACNACCTGTTCCTGTGTATCTGCTA
+
AAAAA#EEEEEEEEEEEEEEEEEEEEEE
@NS500648:585:HK55NAFX2:1:11101:13450:1033 1:N:0:ACAAGGTA
GCCACNACCTGTTCCTGTTGAGGATTTG
+
AAAAA#EEEEEEEEEEEEEEEEEEEEEE
@NS500648:585:HK55NAFX2:1:11101:12961:1039 1:N:0:CGTCCCGT
GCCACNACCTGTTCCTGTTTTCTCTCTG
(metacell) [16:29:53 polivar@aquarius:~/src/chromap_l/chromap-80f75ed_x64-linux] (34512)$
head r2.fq
@NS500648:585:HK55NAFX2:1:11101:9001:1025 2:N:0:ACAAGGTA
NNNNNGCATGCAAAGTCAAAAAACACTNTCATGGTCTTATAATCTGCATTTATTTTTACCTAATNNNCCNNNNGACTCNNNTANNNNTCNTNCANTGATTCNNNTGNTTCCAANNCCNNN
+
#####EEEEEEEEEEEEEEEEEEEEEE#EEEEEEEEEEEEEEEEEEEEAEEEEEEEEEEEEEEE###EE####E<EEE###A/####EE#E#AE#EEE/AA###AA#AAAAA/##<E###
@NS500648:585:HK55NAFX2:1:11101:13450:1033 2:N:0:ACAAGGTA
NNNTGTTATTGTTACATTTTTCTTACAGCCCTTTAACAATCTCATTTCATTTTCATCCGCTTTCGGGGCTGGGTCATGAAGGCCGCAGTCTTAGGTAAGAACTCCACATACATAGTCCCG
+
###AAAEEEEEEE/EEEEEEEAEEEEEEEEEEEEEEEEEEEEEEEEEEE<EEAEEEAEEEEEEE/EEEAAEEEEAEEEEEEEEEEAEEEAEE<AEE<AAA<AAEA6<6AA<EEEAEEEE/
@NS500648:585:HK55NAFX2:1:11101:12961:1039 2:N:0:CGTCCCGT
NNNCAATGGAAGTAAGGGTCCAAACGCAGTTTATTAACAGCTAAGCCAAGCAGGCAAGGGTGAAACTTTGGCAAACGGAATTATGTGGGTAATCCAATAGCACAGTTGTAGTCACAGACT
sed -n '2~4p' r1.fq | wc -l
664948
sed -n '2~4p' r2.fq | wc -l
664948
Edit: The software runs well using the provided fastqs and test ref.
As far as I understand, chromap is able to parse CB from a whitelist and look for it into a specific fastq file (-b). I wonder if it would be the time to create a flexible interface for CB and (possibly) UMIs. For example: in 10x data R2 contains CB and it is as long as required by CB; in SNARE-seq CB is in R1 but the read is longer, including the sections for UMI and a T-tail. Other technologies may introduce CB in the same read with genomic information.
Other tools for scRNA-seq have tackled this before, I'm thinking to bustools, which allows custom architectures.
Chromap is indeed an ultrafast tool. Thanks for developing and this method.
However, I have encountered two problems when I using chromap with scATAC-seq data.
The first one is that when I analyzed the distribution of fragment length, I found a dramatic loss of fragments between 47bp-120bp
The results are shown in these figures.
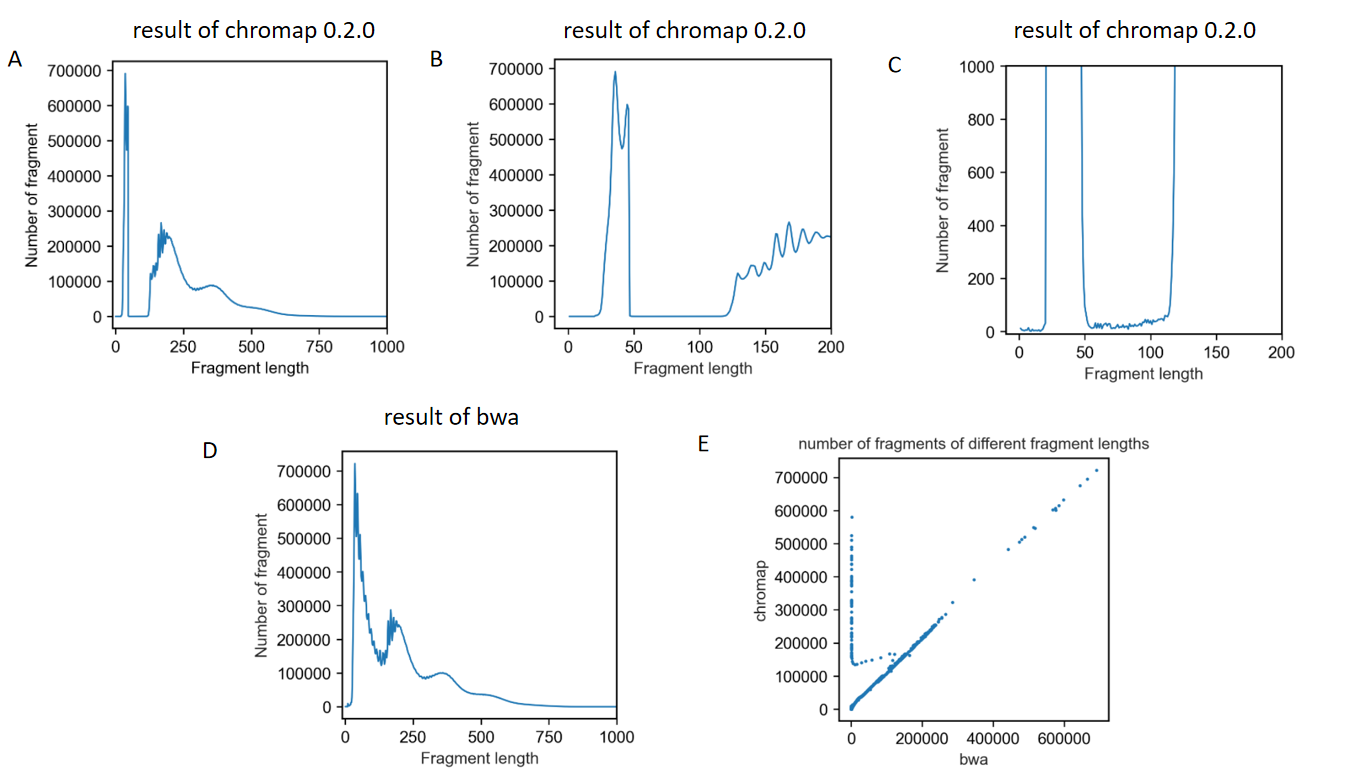
Another problem is that fragments of 0bp existed in the result bed file.

These two problems existed for all data of my different experiments. All these data have 10x-like scATAC-seq structures, using 150bp paired end sequencing.
The command was like this:
chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output} -b ${inputR2} --barcode-whitelist ${whitelist}
I also tried without the barcode fastq, running like this:
chromap --preset atac -x ${index} -r ${fasta} -1 ${inputR1} -2 ${inputR3} -o ${output}
However, the two problems still existed for my data.
When I tried another scATAC-seq dataset with 50bp paired end sequencing, there two problems did not appear.
Hi, I am getting the following error during the index creation of the genome.
I tried this to solve the problem, but failed. #9
And then, used --low-mem, it is the same error.
Build index for the reference.
Kmer length: 17, window size: 7
Reference file: ../GRCh38.primary_assembly.genome.fa
Output file: ./
Loaded all sequences successfully in 20.09s, number of sequences: 194, number of bases: 3099750718.
Collected 739880607 minimizers.
Sorted minimizers.
Kmer size: 17, window size: 7.
Lookup table size: 393121383, # buckets: 536870912, occurrence table size: 448256428, # singletons: 291624179.
Built index successfully in 293.65s.
[M::Statistics] kmer size: 17; skip: 7; #seq: 194
[M::Statistics::3.783] distinct minimizers: 393121383 (74.18% are singletons); average occurrences: 1.882; average spacing: 4.190
chromap: src/index.cc:271: void chromap::Index::Save(): Assertion `index_file != __null' failed.
/public/home/luzhang/.lsbatch/1640139766.46956303: line 8: 17674 Aborted (core dumped) chromap -i -r ../GRCh38.primary_assembly.genome.fa --low-mem -o ./
I'm aligning 10x genomics scATAC-seq to the rheMac10 genome. The sequencing is 50bp for R1 and R2 paired-end sequencing on the Novaseq. The genome was built with k17 and w7 for min frag 30. But the alignment rate is 7% which is too low. For 10x scATAC-seq datasets, how was the genome built to better map fragments as low as 20 or 30 bp after index trimming? Are there any other advice to work with this short of sequencing?
In a different dataset where the sequencing strategy was 100bp for R1 and R2, the same macaque index aligned 91% of the reads.
Mapped all reads in 55348.73s.
Number of reads: 1132465712.
Number of mapped reads: 77938628.
Number of uniquely mapped reads: 72568652.
Number of reads have multi-mappings: 5369976.
Number of candidates: 781477061.
Number of mappings: 77938628.
Number of uni-mappings: 72568652.
Number of multi-mappings: 5369976.
Number of barcodes in whitelist: 2890.
Number of corrected barcodes: 41349117.
Sorted, deduped and outputed mappings in 17.12s.
Hi,
I'm seeing a very low number of mapped reads as can be seen with:
Number of reads: 71940110.
Number of mapped reads: 269214
I'm using the default parameters when running:
/chromap-0.1_x64-linux/chromap -r SpikeIns_Synthetic_New71_AND_hg38.fa -1 RD0175-IC_S36.pe_1.trimmed.fastq.gz -2 RD0175-IC_S36.pe_2.trimmed.fastq.gz -x SpikeIns_Synthetic_New71_AND_hg38_chromap_index --SAM -o RD0175-IC_S36.sam 1>&2
Number of threads: 1
Analyze bulk data.
Won't try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Could this issue be because of "Will remove PCR duplicates at bulk level" ? I haven't set the parameter --remove-pcr-duplicates-at-bulk-level though or any of the --remove-pcr-duplicates parameters.
Also, I haven't specified any barcode (whitelist) file and haven't set the parameter --output-mappings-not-in-whitelist.
Please provide pointers on what could be the issue. Happy to provide any other info as needed. Thanks very much.
❯ chromap --version
0.1-r208
❯ which igv.sh
~/IGV_Linux_2.8.12/igv.sh
When loading the chromap-mapped(0.1-r208) paried-ended bam file to IGV ( version :Linux 2.8.12 ), the forward reads display fine, but the reverse reads showed lots of mismatches bases. By blatting the reverse reads, I found the mapping position was true but the mistake was due to the direction of the reads, I guess chromap didn't give the true sequence information of the reverse reads in the bam file 
Error executing process > 'LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN (scatac_1)'
Caused by:
Process `LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN (scatac_1)` terminated with an error exit status (2)
Command executed:
chromap \
-t 12 \
-r genome.fa \
-x genome.index \
-1 scatac_1_1_val_1.fq.gz \
-2 scatac_1_2_val_2.fq.gz \
-b scatac_1.cellbc.fastq.gz \
--barcode-whitelist hydrop_atac_1_0_whitelist_wadpt.tsv \
-o genome.bed \
-l 2000 --bc-error-threshold 2 --remove-pcr-duplicates --remove-pcr-duplicates-at-cell-level --low-mem --Tn5-shift --BED
cat <<-END_VERSIONS > versions.yml
"LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN":
chromap: $(chromap --version 2>&1)
END_VERSIONS
Command exit status:
2
Command output:
(empty)
Command error:
github.com/sylabs/singularity/internal/app/starter.Master.func2()
RN: Kiinternal/app/starter/master_linux.go:152 +0x3e fp=0xc0001e3f38 sp=0xc0001e3f00 pc=0x73fd8e
github.com/sylabs/singularity/internal/pkg/util/mainthread.Execute.func1()
internal/pkg/util/mainthread/mainthread.go:21 +0x2f fp=0xc0001e3f60 sp=0xc0001e3f38 pc=0x73e59f
main.main()
cmd/starter/main_linux.go:102 +0x5f fp=0xc0001e3f98 sp=0xc0001e3f60 pc=0x915dff
runtime.main()
/camp/apps/eb/software/Go/1.11.4/src/runtime/proc.go:201 +0x207 fp=0xc0001e3fe0 sp=0xc0001e3f98 pc=0x42f8d7
runtime.goexit()
/camp/apps/eb/software/Go/1.11.4/src/runtime/asm_amd64.s:1333 +0x1 fp=0xc0001e3fe8 sp=0xc0001e3fe0 pc=0x45b3f1
goroutine 5 [syscall]:
os/signal.signal_recv(0xaf0720)
/camp/apps/eb/software/Go/1.11.4/src/runtime/sigqueue.go:139 +0x9c
os/signal.loop()
/camp/apps/eb/software/Go/1.11.4/src/os/signal/signal_unix.go:23 +0x22
created by os/signal.init.0
/camp/apps/eb/software/Go/1.11.4/src/os/signal/signal_unix.go:29 +0x41
goroutine 7 [chan receive]:
github.com/sylabs/singularity/internal/pkg/util/mainthread.Execute(0xc0002f6950)
internal/pkg/util/mainthread/mainthread.go:24 +0xb4
github.com/sylabs/singularity/internal/app/starter.Master(0x7, 0x4, 0x11f2, 0xc00000cb60)
internal/app/starter/master_linux.go:151 +0x405
main.startup()
cmd/starter/main_linux.go:75 +0x4b8
created by main.main
cmd/starter/main_linux.go:98 +0x35
rax 0x0
rbx 0x0
rcx 0xffffffffffffffff
rdx 0x0
rdi 0x116e
rsi 0x6
rbp 0xc0001e3ef0
rsp 0xc0001e3eb0
r8 0x0
r9 0x0
r10 0x0
r11 0x206
r12 0xc
r13 0xff
r14 0xae555c
r15 0x0
rip 0x472a9b
rflags 0x206
cs 0x33
fs 0x0
gs 0x0
Hi All,
I am using Hi-C reads, and am testing different -q parameter values. I noticed what seems to be an inconsistency between the manual and the output.
It looks like the default value for -q is 30 based on the manual.
Here's the line:
-q, --MAPQ-threshold INT Min MAPQ in range [0, 60] for mappings to be output [30]
When I run chromap with -q 30, and without any -q options, the resulting pairs files and Hi-C maps look very different. Above the diagonal in the attached picture is with the -q 30 option, and below the diagonal is running chromap without specifying anything for -q. My command is similar to the one in the manual, and follows this general form: chromap --preset hic -x assem.fa.index -r assem.fa -1 R1.fq.gz -2 R2.fq.gz -t 89 -o test.pairs.

So, is the default -q value not 30? What is it? Thank you!
Hi!
Apologies if I have missed this somewhere, and I am trying to process some scATAC-Seq data with chromap, there were more than one barcode and UMI molecular in the data, which there were two link sequence in the barcode reads and barcodes were not continuous.
chromap can process this data?
if so, how about the barcode mode is BC1+Adaptor1+BC2+Adaptor2+BC3+Adaptor3+BC4+UMI+PolyT?
BC1=8bp,BC2=8bp,BC3=8bp,BC4=8bp,UMI=12bp,Adaptor1=30bp,Adaptor2=25bp,Adaptor3=20bp
Thanks
Tao
Chromap looks awesome based on the preprint!! 🥇
If I understand correctly, for chromap to work correctly I would have to set a --min-frag-length so that the index can be built appropriately. How do I estimate this beforehand? I am trying to add chromap to "my" pipeline (seq2science), and I would like for this setting to be set automatically. It generates loads of QC, so could I just look for the smallest read in my sample, and use that? What would you recommend?
Ciao !
On a fresh MAESTRO — release 1.5.1 — installation (Ubuntu 18.04 with Python v3.8.12 installed) I tried to run chromap, chromap --version or chromap -h having immediately Illegal instruction (core dumped) as a result.
I did also try to update chromap with conda install -c bioconda chromap obtaining chromap-0.1.5-h9a82719_0 with no error during installation but, again the Illegal instruction error happened.
What can I do? Thanks
Hello @haowenz:
1. I am wondering how chromap handel Hi-C without knowing the information of restriction enzyme?
2. I am testing chromap with a Hi-C library using combination of two restriction enzymes. And I have generated pairs files. Then can I convert .pairs into .hic or .cool files directly without any filter?
Thanks!
Linhua
Hi all,
I understand SAM output is not optimized and it is discouraged, however I had to test it and I run into some issues.
First of all, output is uncompressed and I couldn't pipe into samtools to obtain a bam file (to save space). However, once SAM is generated I have
$ samtools flagstat test.sam
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
[W::sam_parse1] mapped query cannot have zero coordinate; treated as unmapped
1509410 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
1509401 + 0 mapped (100.00% : N/A)
1509410 + 0 paired in sequencing
754705 + 0 read1
754705 + 0 read2
0 + 0 properly paired (0.00% : N/A)
1509401 + 0 with itself and mate mapped
0 + 0 singletons (0.00% : N/A)
1509401 + 0 with mate mapped to a different chr
1509401 + 0 with mate mapped to a different chr (mapQ>=5)
Strangely, all pairs are mapping on different chromosomes. More interestingly, when I convert to BAM:
$ samtools view -bo test.bam test.sam
[E::bam_write1] Positional data is too large for BAM format
samtools view: writing to "test.bam" failed
but then
$ samtools quickcheck test.bam
$ echo $?
0
which makes it "compliant". But then, again,
$ samtools flagstat test.bam
717631 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
717631 + 0 mapped (100.00% : N/A)
717631 + 0 paired in sequencing
358801 + 0 read1
358830 + 0 read2
0 + 0 properly paired (0.00% : N/A)
717631 + 0 with itself and mate mapped
0 + 0 singletons (0.00% : N/A)
717631 + 0 with mate mapped to a different chr
717631 + 0 with mate mapped to a different chr (mapQ>=5)
Half of the reads disappeared from the alignment
Hi there,
I recently started using chromap on some of my lab's CHIPseq reads and found that the outputted MAPQ scores (for 30-some input files) are all ranging from "0" to "29", with none higher than that. I'm wondering if you could think of a reason why this might be happening. Is there a setting I might have missed/something to do with a parameter that was set on the generation of our reference index file?
Thanks in advance for any help!
Best,
Chris
Hi,
I wonder that if I can use chromap to align the genome re-sequencing data, and is the output BAM file compatible with downstream applications, such as GATK. Thank you very much.
Best regards,
Zheng zhuqing
I output the sam file but failed convert to bam. It said "[E::sam_parse1] CIGAR and query sequence are of different length".
I used custom_read_format version!!!
Hi,
I have tried chromap on public scATAC-seq data without whitelist. If I use the command --barcode-whitelist None, which is supported in STAR, the output bed file is empty. If I generate a whitelist by myself, using all barcodes in fastq file, chromap raised an error as follows :
chromap: src/chromap.cc:5466: void chromap::Chromap::LoadBarcodeWhitelist() [with MappingRecord = chromap::PairedEndMappingWithBarcode]: Assertion `khash_return_code != -1 && khash_return_code != 0' failed.
Aborted (core dumped)
So how to analyze scATAC-seq data without whitelist using chromap?
Best,
Hailin
I have my own scATAC-seq data with barcode 20bp. The error is copy below and with 16G core file:
chromap: src/chromap.cc:3892: void chromap::Chromap::LoadBarcodeWhitelist() [with MappingRecord = chromap::PairedEndMappingWithBarcode]: Assertion `khash_return_code != -1 && khash_return_code != 0' failed.
/opt/gridengine/default/spool/cngb-compute-e05-8/job_scripts/2110610: line 9: 58354 Aborted (core dumped)
Is there anything in particular that needs attention regarding data formatting(read head, barcode length)? The error maybe caused by the data format.
Hi~
I got the following error:
"chromap: src/chromap.cc:4493: void chromap::Chromap::LoadBarcodeWhitelist() [with MappingRecord = chromap::PairedEndMappingWithBarcode]: Assertion `barcode_length == barcode_length_' failed."
--read-format bc:0:19,r1:20:-1
whitelist.txt :CTATGTATAACTATGTATAA 20bp.
I don't know why this error occurs.
Thank you!
Hi, is the fasta file required as an input when aligning along with the index? Please let me know asap. Thanks! Rajat
Hi I used the chip preset to align some paired end reads. The aligned reads look like:
chr1 10099 10287 N 42 -
chr1 16177 16524 N 48 +
chr1 17443 17594 N 44 -
chr1 17475 17662 N 40 +
What do the strands indicate here? The counts of + and - are also not equal (11517045 - and 9372320 +), so they are unlikely to be fragments cut into reads like in tagAlign format.
The maximum size of each line is 2000, which is the default maximum insert length. So what are the strands signifying?
Hi~
Thanks for the useful software! Are you considering adding score and Beto the bed file output from single cell ATAC mode?
Best,
Yumo Song
Hi, thanks for the tool chromap, which looks really promising based on the preprint.
I'm having trouble creating genome index for human. I don't have any problem with the test data, and I successfully created the index of test/ref.fa. However, when I run the following command for human genome:
~/tools/chromap/chromap -i -r ~/reference/homo_sapiens/ucsc/hg38/genome_fa/genome_chrom.fa -o hs_chrom_index
where genome_chrom.fa contains fasta records of human chr1-22, chrX, chrY and chrM. I got the following output and error:
Build index for the reference.
Kmer length: 17, window size: 7
Reference file: /work/bio-chenx/reference/homo_sapiens/ucsc/hg38/genome_fa/genome_chrom.fa
Output file: hs_chrom_index
Loaded all sequences successfully in 4.27s, number of sequences: 25, number of bases: 3088286401.
Collected 737136231 minimizers.
Sorted minimizers.
Kmer size: 17, window size: 7.
Lookup table size: 392946789, # buckets: 536870912, occurrence table size: 445474231, # singletons: 291662000.
Built index successfully in 197.98s.
[M::Statistics] kmer size: 17; skip: 7; #seq: 25
chromap: src/index.cc:19: void chromap::Index::Statistics(uint32_t, const chromap::SequenceBatch&): Assertion `len == reference.GetNumBases()' failed.
Aborted (core dumped)
I tired the pre-compile binary and cloning the github repo and compiling by myself. The same thing happened.
Could you help? Thank you!
Regards,
Xi
Hello.
Is there any limit to the length of chromosomes in chromap?
I'm working on an contact map for genomes with chromosomes greater than 2GB, and some other alignment tools seem to have length limitations. Then I found chromap, so I want to know if it has a limit.
Best wishes.
Hello,
I'm trying to use chromap on ATAC-seq data but I keep getting getting segfaults. I'm using version 0.1.3 (downloaded from [chromap-0.1.3_x64-linux.tar.bz2]) on CentOS 7 with datasets in SRP234892 (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA593838&o=acc_s%3Aa) SRR10597272-9.
System details:
NAME="CentOS Linux"
VERSION="7 (Core)"
ID="centos"
ID_LIKE="rhel fedora"
VERSION_ID="7"
PRETTY_NAME="CentOS Linux 7 (Core)"
ANSI_COLOR="0;31"
CPE_NAME="cpe:/o:centos:centos:7"
HOME_URL="https://www.centos.org/"
BUG_REPORT_URL="https://bugs.centos.org/"
CENTOS_MANTISBT_PROJECT="CentOS-7"
CENTOS_MANTISBT_PROJECT_VERSION="7"
REDHAT_SUPPORT_PRODUCT="centos"
REDHAT_SUPPORT_PRODUCT_VERSION="7"
I also tried on other datasets and ran into the same issue. One of my colleagues has run this version of chromap on our system on a different dataset and that worked fine.
The error I get with chromap-0.1.3_x64-linux.tar.bz2 is as follows:
Will try to remove adapters on 3'.
Will remove PCR duplicates after mapping.
Will remove PCR duplicates at cell level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Perform Tn5 shift.
Output mappings in SAM format.
Reference file: /home/research/vaquerizas/store/genomes/insects/Dmel/6.07/fasta/dmel-all-chromosome-r6.07.fasta
Index file: /home/research/vaquerizas/store/genomes/insects/Dmel/ChromapIndexes/6.07/index
1th read 1 file: fastq/SRR10597272_1.fastq.gz
1th read 2 file: fastq/SRR10597272_2.fastq.gz
Output file: aligned/SRR10597272_chromap.sam
Loaded all sequences successfully in 3.66s, number of sequences: 1870, number of bases: 143726002.
Kmer size: 17, window size: 7.
Lookup table size: 28722375, occurrence table size: 8529222.
Loaded index successfully in 4.69s.
Mapped 500000 read pairs in 26.49s.
/bin/bash: line 1: 190805 Segmentation fault (core dumped) chromap --preset atac -x /home/research/vaquerizas/store/genomes/insects/Dmel/ChromapIndexes/6.07/index -r /home/research/vaquerizas/store/genomes/insects/Dmel/6.07/fasta/dmel-all-chromosome-r6.07.fasta -1 fastq/SRR10597272_1.fastq.gz -2 fastq/SRR10597272_2.fastq.gz --SAM -o aligned/SRR10597272_chromap.sam
Tried again with chromap-0.1.3-asan_x64-linux.tar.bz2 and got:
[nmaziak1@node-head1a fastq]$ chromap-asan --trim-adapters -l 2000 --remove-pcr-duplicates-at-cell-level --Tn5-shift -x /home/research/vaquerizas/store/genomes/insects/Dmel/ChromapIndexes/6.07/index -r /h
ome/research/vaquerizas/store/genomes/insects/Dmel/6.07/fasta/dmel-all-chromosome-r6.07.fasta -1 SRR10597272_1.fastq.gz -2 SRR10597272_2.fastq.gz -o try.bed
Start to map reads.
Parameters: error threshold: 8, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 2000, MAPQ-threshold: 30, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 1
Analyze bulk data.
Will try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at cell level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Perform Tn5 shift.
Output mappings in BED/BEDPE format.
Reference file: /home/research/vaquerizas/store/genomes/insects/Dmel/6.07/fasta/dmel-all-chromosome-r6.07.fasta
Index file: /home/research/vaquerizas/store/genomes/insects/Dmel/ChromapIndexes/6.07/index
1th read 1 file: SRR10597272_1.fastq.gz
1th read 2 file: SRR10597272_2.fastq.gz
Output file: try.bed
Loaded all sequences successfully in 1.93s, number of sequences: 1870, number of bases: 143726002.
Kmer size: 17, window size: 7.
Lookup table size: 28722375, occurrence table size: 8529222.
Loaded index successfully in 3.06s.
Mapped 500000 read pairs in 47.95s.
AddressSanitizer:DEADLYSIGNAL
=================================================================
==64100==ERROR: AddressSanitizer: SEGV on unknown address 0x000000000030 (pc 0x00000077487e bp 0x0000fffffff6 sp 0x7fff184bac00 T0)
==64100==The signal is caused by a READ memory access.
==64100==Hint: address points to the zero page.
#0 0x77487e in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::VerifyCandidatesOnOneDirection(chromap::Direction, chromap::SequenceBatch const&, unsigned int, chromap::SequenceBatch const&, std::vector<chromap::Candidate, std::allocator<chromap::Candidate> > const&, std::vector<std::pair<int, unsigned long>, std::allocator<std::pair<int, unsigned long> > >*, std::vector<int, std::allocator<int> >*, int*, int*, int*, int*) (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x77487e)
#1 0x7ef445 in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::VerifyCandidates(chromap::SequenceBatch const&, unsigned int, chromap::SequenceBatch const&, std::vector<std::pair<unsigned long, unsigned long>, std::allocator<std::pair<unsigned long, unsigned long> > > const&, std::vector<chromap::Candidate, std::allocator<chromap::Candidate> > const&, std::vector<chromap::Candidate, std::allocator<chromap::Candidate> > const&, std::vector<std::pair<int, unsigned long>, std::allocator<std::pair<int, unsigned long> > >*, std::vector<int, std::allocator<int> >*, std::vector<std::pair<int, unsigned long>, std::allocator<std::pair<int, unsigned long> > >*, std::vector<int, std::allocator<int> >*, int*, int*, int*, int*) (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x7ef445)
#2 0x8271da in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::MapPairedEndReads() [clone ._omp_fn.2] (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x8271da)
#3 0x9fc635 in GOMP_taskloop (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x9fc635)
#4 0x6b75e9 in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::MapPairedEndReads() [clone ._omp_fn.0] (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x6b75e9)
#5 0x9f68f1 in GOMP_parallel (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x9f68f1)
#6 0x8aab30 in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::MapPairedEndReads() (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x8aab30)
#7 0x5ebc16 in chromap::ChromapDriver::ParseArgsAndRun(int, char**) (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x5ebc16)
#8 0x41bdbf in main (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x41bdbf)
#9 0x7f10f35bc554 in __libc_start_main (/lib64/libc.so.6+0x22554)
#10 0x41ea1e (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x41ea1e)AddressSanitizer can not provide additional info.
SUMMARY: AddressSanitizer: SEGV (/home/nmaziak1/software/chromap-0.1.3-asan_x64-linux/chromap-asan+0x77487e) in chromap::Chromap<chromap::PairedEndMappingWithoutBarcode>::VerifyCandidatesOnOneDirection(chromap::Direction, chromap::SequenceBatch const&, unsigned int, chromap::SequenceBatch const&, std::vector<chromap::Candidate, std::allocator<chromap::Candidate> > const&, std::vector<std::pair<int, unsigned long>, std::allocator<std::pair<int, unsigned long> > >*, std::vector<int, std::allocator<int> >*, int*, int*, int*, int*)
==64100==ABORTING
As far as I can tell the indexing has been fine. Also of note, without the option --SAM or --trim-adapters it will run for a little longer until it crashes into the same error. Last, I accidentally fed it the same read pair once and it actually went through it just fine (of course telling me nothing paired). The --low-mem option doesn’t seem to change anything.
I have had general issues with ATAC-seq datasets from drosophila (no issues however with ChIP/RNAseq/or GROseq), not sure why but just for reference, the code I run to get them is : fastq-dump --gzip --split-3 --readids -B --skip-technical --clip SRR10597272
Thank you!
Noura
Hello,
Thanks for developing Chromap. I was trying to install it using this command: conda install -c bioconda chromap. However, I got the folllowing errors:
Collecting package metadata (current_repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: failed with repodata from current_repodata.json, will retry with next repodata source.
Collecting package metadata (repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: |
Found conflicts! Looking for incompatible packages.
Do you know how to solve this issue?
Best,
The sam file generated by -SAM option didn't contain sequence information.
The command used for alignment:
chromap --preset chip \ -x ${chromapindex} \ -r ${genome} \ -1 ${outdir}//0-clean_data/paired/${key}_val_1.fq \ -2 ${outdir}//0-clean_data/paired/${key}_val_2.fq \ --SAM -t ${thread} -q 1 \ -o ${outdir}//1-chromap/sam/${key}_q1.sam
The sam output:
$ samtools view input_1_q1.sam|head -n 10
E00517:731:HG57TCCX2:5:1222:27042:52731 97 chr1 16203 50 150M * 0 0 * * NM:i:0 MD:Z:150
E00517:731:HG57TCCX2:5:1218:1905:44310 97 chr1 16352 45 147M * 0 0 * * NM:i:0 MD:Z:147
E00517:731:HG57TCCX2:5:1218:1905:44310 145 chr1 16247 45 148M * 0 0 * * NM:i:5 MD:Z:20G5G88A14A15A1
E00517:731:HG57TCCX2:5:1222:27042:52731 145 chr1 16283 50 150M * 0 0 * * NM:i:0 MD:Z:150
E00517:731:HG57TCCX2:5:1212:29193:35819 97 chr1 16806 18 149M * 0 0 * * NM:i:0 MD:Z:149
E00517:731:HG57TCCX2:5:1212:29193:35819 145 chr1 16688 18 150M * 0 0 * * NM:i:3 MD:Z:8C4C46T89
E00517:731:HG57TCCX2:5:2108:20172:14002 161 chr1 17089 46 150M * 0 0 * * NM:i:0 MD:Z:150
E00517:731:HG57TCCX2:5:2108:20172:14002 81 chr1 17302 46 150M * 0 0 * * NM:i:0 MD:Z:150
E00517:731:HG57TCCX2:5:1212:3224:52994 161 chr1 19167 49 145M * 0 0 * * NM:i:0 MD:Z:145`
Could the information be included?
Thank you for this excellent tool!
I cannot find any reference to how you remove PCR duplicates. Can you confirm it is similar to the 10x "new" method? If you do not have a barcode whitelist - how do you remove duplicates at the cell level?
https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/algorithms/overview
I think the documentation could be extended to include more complete descriptions of your algorithms in general.
Hi, I got the following error when creating the wheat genome index
Build index for the reference.
Kmer length: 17, window size: 7
Reference file: 161010_Chinese_Spring_v1.0_pseudomolecules.fasta
Output file: wheat_chromap_chipseq.index
Loaded all sequences successfully in 59.10s, number of sequences: 22, number of bases: 14547261565.
Collected 3566471558 minimizers.
Sorted minimizers.
chromap: src/index.cc:134: void chromap::Index::Construct(uint32_t, const chromap::SequenceBatch&): Assertion `num_minimizers != 0 && num_minimizers <= INT_MAX' failed
Because the reference genome of wheat is very large(~ 17 Gb), tools that can improve the speed of mapping are valuable for wheat.
The cause of the error is most likely that the wheat reference genome of wheat is too large. This problem is also common when using other software for wheat genomes. However, BWA build index does not report errors.
Wheat genome download site: http://plants.ensembl.org/Triticum_aestivum/Info/Index
For 10x data, cellranger-atac/cellranger-arc make their whitelist in .tsv.gz format. It would be convenient if chromap can take gzipped file as input here (similar to fastq file).
Thanks for developing such a useful tool!
Hi, is there any way to force chromap to output all the unmapped and multi-mapped reads?
And how are they identified? It was mentioned in issue 20 that multi-mapped reads are assigned mapq=0, how about unmapped?
The reason I am asking is because I have some downstream QC which will report the unmapped and multi-mapped numbers in a SAM/BAM file.
#20
Hi,
We have mapped our illumina short reads to our genome using typical commands with --SAM of chromap.

But I failed to find the tag like "NH:i:" or "XT:A:U" at the 12th line of our sam output.

Can I extract unique mapped reads from the sam output of chromap?
Program will not build an index on chr20 of hg38 - I have tried many different k and w values but it doesn't seem to work
Command executed:
chromap \
-i \
-r hg38-chr20.fa \
-o hg38-chr20.index \
-k 17 -w 7
cat <<-END_VERSIONS > versions.yml
"LUSLAB_CUSTOMSC:CUSTOMSC:PREPARE_GENOME:CHROMAP_INDEX":
chromap: $(chromap --version 2>&1)
END_VERSIONS
Command exit status:
134
Command output:
(empty)
Command error:
Build index for the reference.
Kmer length: 17, window size: 7
Reference file: hg38-chr20.fa
Output file: hg38-chr20.index
Loaded all sequences successfully in 0.21s, number of sequences: 1, number of bases: 1632598.
Collected 0 minimizers.
Sorted minimizers.
chromap: src/index.cc:179: void chromap::Index::Construct(uint32_t, const chromap::SequenceBatch&): Assertion `num_minimizers != 0 && num_minimizers <= 0x7fffffff' failed.
.command.sh: line 6: 29 Aborted chromap -i -r hg38-chr20.fa -o hg38-chr20.index -k 17 -w 7
Hi there,
When running chromap with the following: chromap --preset chip -x genome.index -r genome.fa -1 SPT5_INPUT_REP1_T1_1_val_1.fq.gz -2 SPT5_INPUT_REP1_T1_2_val_2.fq.gz -o SPT5_INPUT_REP1_T1.bed, I get the process kill, as you can see below:
Preset parameters for ChIP-seq are used.
Start to map reads.
Parameters: error threshold: 8, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 2000, MAPQ-threshold: 30, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 1
Analyze bulk data.
Won't try to remove adapters on 3'.
Will remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Output mappings in BED/BEDPE format.
Reference file: genome.fa
Index file: genome.index
1th read 1 file: SPT5_INPUT_REP1_T1_1_val_1.fq.gz
1th read 2 file: SPT5_INPUT_REP1_T1_2_val_2.fq.gz
Output file: SPT5_INPUT_REP1_T1.bed
Loaded all sequences successfully in 0.02s, number of sequences: 17, number of bases: 12157105.
Kmer size: 17, window size: 7.
Lookup table size: 2857826, occurrence table size: 243803.
Loaded index successfully in 0.03s.
Mapped 99321 read pairs in 0.67s.
Mapped all reads in 0.92s.
Number of reads: 198642.
Number of mapped reads: 187220.
Number of uniquely mapped reads: 165306.
Number of reads have multi-mappings: 21914.
Number of candidates: 403416.
Number of mappings: 187220.
Number of uni-mappings: 165306.
Number of multi-mappings: 21914.
KilledI reproduced the error using the test data:
$ chromap --preset chip -x ref.index -r ref.fa -1 read1.fq -2 read2.fq -o test.bed
Preset parameters for ChIP-seq are used.
Start to map reads.
Parameters: error threshold: 8, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 2000, MAPQ-threshold: 30, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 1
Analyze bulk data.
Won't try to remove adapters on 3'.
Will remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Output mappings in BED/BEDPE format.
Reference file: ref.fa
Index file: ref.index
1th read 1 file: read1.fq
1th read 2 file: read2.fq
Output file: test.bed
Loaded all sequences successfully in 0.00s, number of sequences: 1, number of bases: 100000.
Kmer size: 17, window size: 7.
Lookup table size: 25079, occurrence table size: 0.
Loaded index successfully in 0.00s.
Mapped 10 read pairs in 0.00s.
Mapped all reads in 0.00s.
Number of reads: 20.
Number of mapped reads: 20.
Number of uniquely mapped reads: 20.
Number of reads have multi-mappings: 0.
Number of candidates: 20.
Number of mappings: 20.
Number of uni-mappings: 20.
Number of multi-mappings: 0.
KilledHowever, if I just use one of the fastq files, both with the real and the test data, it works e.g. with the test data chromap --preset chip -x ref.index -r ref.fa -1 read1.fq -o test.bed
I am running it using the last version of chromap 0.1.5-r302 in a conda environment in Ubuntu 18.04.6.
Thanks a lot!
Hi,
I've tried Chromap for mapping Hi-C reads for scaffolding with SALSA2.
I generated a bed output with:
chromap --preset hic -x index -r cfish.ref.fa -1 SL01_R1.fq -2 SL01_R2.fq --BED -o ctfish.aln.bed
However, the output bed file is not compatible with the expected bed format in SALSA2.
A workaround is to generate a SAM output with chromap --SAM first, then convert the sam/bam file to bed format as expected by SALSA2.
Best,
Julien
Hi all,
I'm testing chromap and I'd say I'm impressed by the performances. In addition to the current options, would you consider the possibility to output counts over given intervals? Catalogs of epigenetic features are available and it would be interesting to quantify known annotations.
Sorry to submit so many errors!
Error executing process > 'LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN (scatac_1)'
Caused by:
Process `LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN (scatac_1)` terminated with an error exit status (134)
Command executed:
chromap \
-t 4 \
-r hg38-chr20.fa \
-x hg38-chr20.index \
-1 scatac_1_1_val_1.fq.gz \
-2 scatac_1_2_val_2.fq.gz \
-b scatac_1.cellbc.fastq.gz \
--barcode-whitelist hydrop_atac_1_0_whitelist_wadpt.tsv \
-o hg38-chr20.bed \
--preset atac
cat <<-END_VERSIONS > versions.yml
"LUSLAB_CUSTOMSC:CUSTOMSC:CHROMAP_ALIGN":
chromap: $(chromap --version 2>&1)
END_VERSIONS
Command exit status:
134
Command output:
(empty)
Command error:
Preset parameters for ATAC-seq/scATAC-seq are used.
Start to map reads.
Parameters: error threshold: 8, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 2000, MAPQ-threshold: 30, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 4
Analyze single-cell data.
Will try to remove adapters on 3'.
Will remove PCR duplicates after mapping.
Will remove PCR duplicates at cell level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Perform Tn5 shift.
Output mappings in BED/BEDPE format.
Reference file: hg38-chr20.fa
Index file: hg38-chr20.index
1th read 1 file: scatac_1_1_val_1.fq.gz
1th read 2 file: scatac_1_2_val_2.fq.gz
1th cell barcode file: scatac_1.cellbc.fastq.gz
Cell barcode whitelist file: hydrop_atac_1_0_whitelist_wadpt.tsv
Output file: hg38-chr20.bed
Loaded all sequences successfully in 0.77s, number of sequences: 1, number of bases: 64444167.
Kmer size: 17, window size: 7.
Lookup table size: 12847197, occurrence table size: 3782818.
Loaded index successfully in 3.01s.
chromap: src/chromap.cc:4804: void chromap::Chromap<MappingRecord>::LoadBarcodeWhitelist() [with MappingRecord = chromap::PairedEndMappingWithBarcode]: Assertion `khash_return_code != -1 && khash_return_code != 0' failed.
.command.sh: line 11: 33 Aborted chromap -t 4 -r hg38-chr20.fa -x hg38-chr20.index -1 scatac_1_1_val_1.fq.gz -2 scatac_1_2_val_2.fq.gz -b scatac_1.cellbc.fastq.gz --barcode-whitelist hydrop_atac_1_0_whitelist_wadpt.tsv -o hg38-chr20.bed --preset atac
Hi,
Thanks for the great software.
I've been running chromap (-preset atac -t 8, otherwise default params) on a lot of samples from the literature, but I'm getting 'core dumped' errors on a small fraction of them, happening right after the "Loaded index successfully" message (hence seems different from #37 ).
One such example is the run SRR4269915 from the SRA (the read length is not uniform across reads in this sample).
When I use the version installed from conda (0.1.3-r256), the error I get is:
25736 Aborted (core dumped)
free(): invalid pointer
When I use the version from the repo (0.1.3-r257; self-built), the error I get is at the same spot but slightly different:
25569 Aborted (core dumped)
chromap: malloc.c:2401: sysmalloc: Assertion `(old_top == initial_top (av) && old_size == 0) || ((unsigned long) (old_size) >= MINSIZE && prev_inuse (old_top) && ((unsigned long) old_end & (pagesize - 1)) == 0)' failed.
I thought maybe there was something wrong with the fastq files, but I tried aligned with bowtie2 and everything went smoothly.
The index was created with default params and works fine for most samples.
(Running on ubuntu 18.04)
When running chromap a lot of information is presented, detailing the options used for the run. I understand that some values can be selected using the --preset option but the user is limited to the chip/atac/hic choices.
Will you expose all options to the user in the future?
Hi,
I want compared the difference between two haplotypes. Can chromap directly alignment two haplotypes? If I want to get alignmet files of two haplotypes precisely. How to get it?
Hello,
I have 50 pair end fastq files, can I input all together to chromap.
Thanks and regards
Sandip
Hello, I get the following compilation errors when make on our CentOS7 HPC, can you help me out here?
Here is the log: https://uni-muenster.sciebo.de/s/hMcvWZgEnIKOo0S
...and here the output from the screen:
https://gist.github.com/ATpoint/a3e0d5eb319895ffb4d894667901afd1
Hi!
I have a quick question regarding PCR duplicates in Hi-C preset, does PCR duplicates get removed already in the output pairs (4DN format)? Could not find this information in the manual page.
Thank you so much!
Jim
Hi,
I've been trying to use chromap for Hi-C data. It is very convenient and super fast!
I have no problem for most Hi-C dataset, however I encountered an "bad_alloc" issue for ultra-deep Hi-C. (more than 1 billion reads). I attached my command line and log below, do you have any idea about solving this issue?
Many thanks
Jim
CPU: 80 threads, memory: 125GB.
command: chromap --preset hic -x chromap-mm10 -r mm10.fa -1 SRR9906313_GSM4010832_HiC_Retina_Adult-Rep1_Mus_musculus_Hi-C_1.fastq.gz -2 SRR9906313_GSM4010832_HiC_Retina_Adult-Rep1_Mus_musculus_Hi-C_2.fastq.gz -o SRR9906313_HiC_Retina_Adult-Rep1_Hi-C.chromap.pairs -t 64
log :
"Mapped all reads in 6054.02s.
Number of reads: 2866604954.
Number of mapped reads: 2582847256.
Number of uniquely mapped reads: 2315287482.
Number of reads have multi-mappings: 267559774.
Number of candidates: 109967150601.
Number of mappings: 2582847256.
Number of uni-mappings: 2315287482.
Number of multi-mappings: 267559774.
terminate called after throwing an instance of 'std::bad_alloc'
what(): std::bad_alloc"
The genome is about 3G and come up with Segmentation fault (core dumped)
cmd: chromap --preset hic -x reference/genome.index --SAM -1 fastq/HIC508_raw_R1.fastq.gz -2 fastq/HIC508_raw_R2.fastq.gz -r reference/genome.fa -t 20 -o hic.sam
Preset parameters for Hi-C are used.
Start to map reads.
Parameters: error threshold: 4, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 1000, MAPQ-threshold: 1, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 20
Analyze bulk data.
Won't try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Allow split alignment.
Output mappings in SAM format.
Reference file: reference/genome.fa
Index file: reference/genome.index
1th read 1 file: fastq/HIC508_raw_R1.fastq.gz
1th read 2 file: fastq/HIC508_raw_R2.fastq.gz
Output file: hic.sam
Loaded all sequences successfully in 4.68s, number of sequences: 291, number of bases: 3047273682.
Kmer size: 17, window size: 7.
Lookup table size: 285958605, occurrence table size: 566349725.
Loaded index successfully in 8.29s.
Mapped 500000 read pairs in 11.97s.
Mapped 500000 read pairs in 7.75s.
Mapped 500000 read pairs in 8.23s.
Mapped 500000 read pairs in 7.61s.
Mapped 500000 read pairs in 7.69s.
Mapped 500000 read pairs in 6.89s.
Mapped 500000 read pairs in 6.80s.
Mapped 500000 read pairs in 38.93s.
Mapped 500000 read pairs in 7.27s.
Mapped 500000 read pairs in 6.59s.
Mapped 500000 read pairs in 6.84s.
Mapped 500000 read pairs in 6.91s.
Mapped 500000 read pairs in 6.99s.
Mapped 500000 read pairs in 6.80s.
Mapped 500000 read pairs in 61.25s.
Mapped 500000 read pairs in 7.04s.
Mapped 500000 read pairs in 6.70s.
Mapped 500000 read pairs in 6.60s.
Mapped 500000 read pairs in 6.54s.
Mapped 500000 read pairs in 6.55s.
Mapped 500000 read pairs in 6.45s.
Mapped 500000 read pairs in 53.85s.
Mapped 500000 read pairs in 6.85s.
Mapped 500000 read pairs in 6.44s.
Mapped 500000 read pairs in 6.32s.
Mapped 500000 read pairs in 6.24s.
Mapped 500000 read pairs in 5.89s.
Mapped 500000 read pairs in 6.31s.
Mapped 500000 read pairs in 45.04s.
Mapped 500000 read pairs in 7.13s.
Mapped 500000 read pairs in 6.83s.
Mapped 500000 read pairs in 6.72s.
Mapped 500000 read pairs in 6.88s.
Mapped 500000 read pairs in 6.82s.
Mapped 500000 read pairs in 6.98s.
Mapped 500000 read pairs in 36.04s.
Mapped 500000 read pairs in 7.41s.
Mapped 500000 read pairs in 6.97s.
Mapped 500000 read pairs in 6.87s.
Mapped 500000 read pairs in 6.77s.
Mapped 500000 read pairs in 6.76s.
Mapped 500000 read pairs in 6.64s.
Mapped 500000 read pairs in 38.23s.
Segmentation fault (core dumped)
I'm mapping some scATAC-seq data using chromap with the following command:
chromap --preset atac \
-x hg38.fna.chromap \
-r hg38.fna.gz \
-1 scATAC_R1.fastq.gz \
-2 scATAC_R3.fastq.gz \
-o mapped/sample.bed \
-b scATAC_R2.fastq.gz \
--barcode-whitelist data/737K-cratac-v1_revcomp.txt \
--read-format bc:15:-1 \
-t 6
After updating to v0.1.5 I get the following error: Less than 5% barcodes can be found or corrected based on the barcode whitelist. Downgrading back to 0.1.3 (what I used previously) with the same command runs without error.
Hi there,
I am trying to run this pipeline with a scATAC-seq dataset generated with 10X chromium.
So, from my understanding based on the tutorial you provide, for -b, the R2 file should be the one to use which contains the information of the CBs. However, for the sequencing file R1, I am not quite sure where to put it.
Here is the code I used after indexing,
chromap --preset atac -x index -r $reference -1 ATACLib_S1_L001_R1_001.fastq.gz -o aln.bed -b ATACLib_S1_L001_R2_001.fastq.gz --barcode-whitelist $work_path/737K-cratac-v1.txt
However, I got an error like :
Start to map reads.
Parameters: error threshold: 8, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 2000, MAPQ-threshold: 30, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90.
Is there any suggestion on how to make this work?
Cheers
Hi, @haowenz
I use the chromap to mapping HiC reads to a plant genome for scaffolding. But it fail when I set the output to /dev/stdout. It throw the error
chromap: src/output_tools.cc:348: void chromap::OutputTools<MappingRecord>::OutputTempMapping(const string&, uint32_t, const std::vector<std::vector<MappingRecord> >&)
[with MappingRecord = chromap::SAMMapping; std::string = std::__cxx11::basic_string<char>; uint32_t = unsigned int]: Assertion `temp_mapping_output_file != __null' failed.When I try to ouptut to a file M32.temp.sam then covert to the bam format, the chromap run successfully but the samtools throw the error
[E::sam_parse1] CIGAR and query sequence are of different length
[W::sam_read1] Parse error at line 20315
[main_samview] truncated file.
# line 20315
A00234:853:HTT5VDSX2:3:2675:19777:22623 179 utg000001l 38590 9 104M * 0 0 ACAGTCCAAAATCAGTCGTTTTTGCTGGAAACTAACCTTGAACCAATCACACATGAGCAAGAATGCTCTGATACCAATTGTAGGTCCCTCTGGGTTGGATCATCCTTAATTCGTTGCTTACTTCATACCACCAGGTTGTTATGAACTATA FF::FFFFFFFFFFFFFFFFFFF:F:FFFFF::FFFF:FF:FFFFFF:F:FFF,,F::FFFFFFFFFFFFFFFFF:FF:FFFFFFFFF:F,FFFFFFFFFFFF,FFFFF:F,FFF,FFFFFFF:FFFFFFF,FF,FFF:FFFF:F::FFF NM:i:2 MD:Z:T1A101
Here is the command I use
chromap -i -r M32.contigs.fa -o M32.index
#stdout output
chromap --preset hic -x M32.index -r assembly.fa -t 12 -q 30 -1 M32.Clean.R1.part_001.fq.gz -2 M32.Clean.R2.part_001.fq.gz --SAM -o /dev/stdout | samtools view -t 12 -Shb -o M32.chromap.bam
#sam file
chromap --preset hic -x M32.index -r assembly.fa -t 12 -1 M32.Clean.R1.part_001.fq.gz -2 M32.Clean.R2.part_001.fq.gz --SAM -o M32.temp.sam
samtools view -t 12 -Shb -o M32.chromap.bam M32.temp.sam
log
# stdout
Preset parameters for Hi-C are used.
Start to map reads.
Parameters: error threshold: 4, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 1000, MAPQ-threshold: 30, min-read-length: $
0, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 12
Analyze bulk data.
Won't try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Allow split alignment.
Output mappings in SAM format.
Reference file: assembly.fa
Index file: M32.index
1th read 1 file: M32.Clean.R1.part_001.fq.gz
1th read 2 file: M32.Clean.R2.part_001.fq.gz
Output file: /dev/stdout
Loaded all sequences successfully in 6.41s, number of sequences: 20307, number of bases: 4462388593.
Kmer size: 17, window size: 7.
Lookup table size: 185123217, occurrence table size: 1044627066.
Loaded index successfully in 30.42s.
Mapped 500000 read pairs in 88.24s.
Mapped 500000 read pairs in 33.60s.
Mapped 500000 read pairs in 29.44s.
Mapped 500000 read pairs in 30.84s.
chromap: src/output_tools.cc:348: void chromap::OutputTools<MappingRecord>::OutputTempMapping(const string&, uint32_t, const std::vector<std::vector<MappingRecord> >&)
[with MappingRecord = chromap::SAMMapping; std::string = std::__cxx11::basic_string<char>; uint32_t = unsigned int]: Assertion `temp_mapping_output_file != __null' fa
iled.sam file log
Preset parameters for Hi-C are used.
Start to map reads.
Parameters: error threshold: 4, min-num-seeds: 2, max-seed-frequency: 500,1000, max-num-best-mappings: 1, max-insert-size: 1000, MAPQ-threshold: 1, min-read-length: 30, bc-error-threshold: 1, bc-probability-threshold: 0.90
Number of threads: 12
Analyze bulk data.
Won't try to remove adapters on 3'.
Won't remove PCR duplicates after mapping.
Will remove PCR duplicates at bulk level.
Won't allocate multi-mappings after mapping.
Only output unique mappings after mapping.
Only output mappings of which barcodes are in whitelist.
Allow split alignment.
Output mappings in SAM format.
Reference file: assembly.fa
Index file: M32.index
1th read 1 file: M32.Clean.R1.part_001.fq.gz
1th read 2 file: M32.Clean.R2.part_001.fq.gz
Output file: M32.temp.sam
Loaded all sequences successfully in 34.30s, number of sequences: 20307, number of bases: 4462388593.
Kmer size: 17, window size: 7.
Lookup table size: 185123217, occurrence table size: 1044627066.
Loaded index successfully in 97.68s.
Mapped 500000 read pairs in 78.43s.
Mapped 500000 read pairs in 30.17s.
Mapped 500000 read pairs in 26.05s.
Mapped 500000 read pairs in 27.59s.
Mapped 500000 read pairs in 30.56s.
Mapped 500000 read pairs in 23.35s.
Mapped 500000 read pairs in 22.57s.
Mapped 500000 read pairs in 22.78s.
Mapped 500000 read pairs in 27.67s.
Mapped 500000 read pairs in 21.89s.
Mapped 500000 read pairs in 21.94s.
Mapped 500000 read pairs in 22.07s.
Mapped 500000 read pairs in 28.24s.
Mapped 500000 read pairs in 22.09s.
Mapped 500000 read pairs in 21.25s.
Mapped 500000 read pairs in 22.55s.
Mapped 500000 read pairs in 26.80s.
Mapped 500000 read pairs in 22.42s.
Mapped 500000 read pairs in 21.73s.
Mapped 500000 read pairs in 20.30s.
Mapped 500000 read pairs in 25.67s.
Mapped 500000 read pairs in 21.24s.
Mapped 500000 read pairs in 22.29s.
Mapped 500000 read pairs in 22.39s.
Mapped 496659 read pairs in 25.47s.
Mapped all reads in 660.55s.
Number of reads: 24993318.
Number of mapped reads: 21819532.
Number of uniquely mapped reads: 9274894.
Number of reads have multi-mappings: 12544638.
Number of candidates: 7360165941.
Number of mappings: 21819532.
Number of uni-mappings: 9274894.
Number of multi-mappings: 12544638.
Sorted and outputed mappings in 64.57s.
# uni-mappings: 17821250, # multi-mappings: 3998242, total: 21819492.
Number of output mappings (passed filters): 6701518
Total time: 873.62s.
[E::sam_parse1] CIGAR and query sequence are of different length
[W::sam_read1] Parse error at line 20315
[main_samview] truncated file.
# line 20315
A00234:853:HTT5VDSX2:3:2675:19777:22623 179 utg000001l 38590 9 104M * 0 0 ACAGTCCAAAATCAGTCGTTTTTGCTGGAAACTAACCTTGAACCAATCACACATGAGCAAGAATGCTCTGATACCAATTGTAGGTCCCTCTGGGTTGGATCATCCTTAATTCGTTGCTTACTTCATACCACCAGGTTGTTATGAACTATA FF::FFFFFFFFFFFFFFFFFFF:F:FFFFF::FFFF:FF:FFFFFF:F:FFF,,F::FFFFFFFFFFFFFFFFF:FF:FFFFFFFFF:F,FFFFFFFFFFFF,FFFFF:F,FFF,FFFFFFF:FFFFFFF,FF,FFF:FFFF:F::FFF NM:i:2 MD:Z:T1A101A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.