Comments (8)
 mourisl
commented on August 24, 2024
mourisl
commented on August 24, 2024
Thanks for sharing your test results with us. Indeed, the length distribution change is too sharp, and the fragment length 0 should not happen either. Is the data publicly available? If it is not, could you please share a few hundreds of the paired-end sequence data with us (no need for the barcode file)? We will look into this issue. Thank you.
from chromap.
 loraince
commented on August 24, 2024
loraince
commented on August 24, 2024
Thank you for your reply.
I uploaded 100,000 read pairs of my paired fastq here without barcode file:
https://github.com/loraince/miscellaneous/blob/main/ATAC_100k.zip
This data contains both human cells and mouse cells.
When mapping to GRCh38, one fragment with length 0 appeared at chr5 134927308 134927308.
Here are the distribution of fragment length.
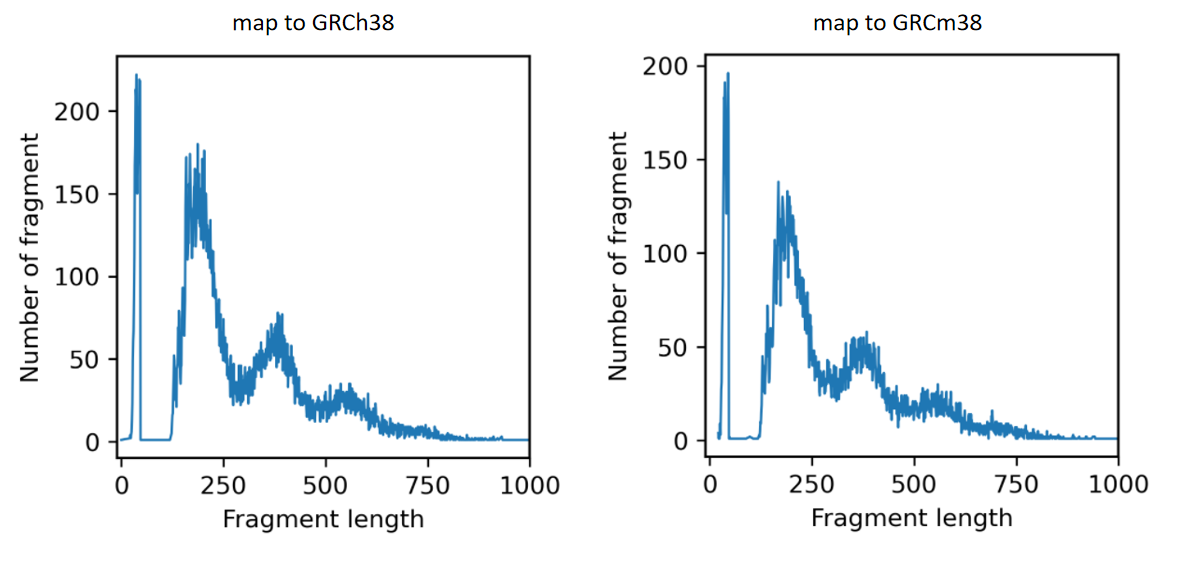
It is worth noting that ATAC_100k_1.fq has 150 bp for each read, while ATAC_100k_2.fq has 55 bp for each read. That is because the barcode is a part of the original R2 file and I split it to get the barcode file and a truncated ATAC_100k_2.fq. I guess the unequal length of R1.fq and R2.fq may cause the problem.
from chromap.
mourisl
commented on August 24, 2024
Thanks for sharing the data! We will look into this issue.
from chromap.
 haowenz
commented on August 24, 2024
haowenz
commented on August 24, 2024
This issue should get fixed in the v0.2.2. You can try it.
from chromap.
loraince
commented on August 24, 2024
Thank you for the updation. I tried with the v0.2.2 and the distribution of fragment length is correct now.
However, the fragments with length 0 still exist. For example, when mapping the small dataset above to the human genome, one fragment was recorded with 0 bp at chr5 134927308 134927308. I am not sure whether you can reproduce this. If not, maybe the difference is between the reference genome. I am unsing human genome from http://cistrome.org/~galib/MAESTRO/references/scATAC/Refdata_scATAC_MAESTRO_GRCh38_1.1.0.tar.gz
By the way, could you please help me with two other questions?
- I find that --trim-adapters is true in --preset atac, while it is not used in --preset chip. I wonder why not trim adapters by default when it comes to chip-seq mode.
- Sometimes we need a BAM file for the downstream analysis. Is there any way to output a BAM file?
from chromap.
haowenz
commented on August 24, 2024
Thank you for the updation. I tried with the v0.2.2 and the distribution of fragment length is correct now.
However, the fragments with length 0 still exist. For example, when mapping the small dataset above to the human genome, one fragment was recorded with 0 bp at chr5 134927308 134927308. I am not sure whether you can reproduce this. If not, maybe the difference is between the reference genome. I am unsing human genome from http://cistrome.org/~galib/MAESTRO/references/scATAC/Refdata_scATAC_MAESTRO_GRCh38_1.1.0.tar.gz
Thank you for testing it again. We will check what happened.
By the way, could you please help me with two other questions?
- I find that --trim-adapters is true in --preset atac, while it is not used in --preset chip. I wonder why not trim adapters by default when it comes to chip-seq mode.
The adapter trimming we have, if you had a look at the paper, since we don't require user to input the adapter sequences, it only trims when are adapters on 3' end. In this case, the two ends of the read pair have an overlap in the middle which is the true fragment (see the last supplementary figure). Our method can detect this overlap and trim the adapters on 3' end. Note that this case happens a lot for scATAC-seq since the fragment length of scATAC-seq can frequently be short like 30-50bp (shorter than read length). On the other hand, for ChIP-seq, the fragment length is usually long. So we don't turn it on by default. But you can turn it on and see how much difference you would observe. In most cases, the difference on ChIP-seq should be very minor.
- Sometimes we need a BAM file for the downstream analysis. Is there any way to output a BAM file?
Currently, Chromap cannot output BAM directly. One way to get BAM is to output mappings in SAM and then convert it to BAM using samtools, just like the way you get BAM when using other aligners (e.g., BWA-MEM). The initial goal of designing Chromap is to bypass SAM and generate fragment files directly, which would save a lot of time. That's why we haven't put much effort on generating SAM or even BAM. Well, we do plan to add BAM support. But it would require a lot of works and it won't happen very soon in the future.
from chromap.
mourisl
commented on August 24, 2024
Hi @loraince, the fragment length 0 alignments seem to be chimeric reads or other sequencing artifacts. The example you mention should be the read: A00253:575:HWWVTDSX2:4:1260:28691:24314, where BWA-MEM aligned it on chrM and also creates a very short fragment size (9). It is by chance that Chromap selects another alignment coordinate with fragment 0. The downstream analysis tool usually filters such short fragments, so I don't think you need to worry about the case.
from chromap.
loraince
commented on August 24, 2024
Thank you both so much for such prompt and detailed replies. My questions have been sufficiently addressed.
from chromap.
Related Issues (20)
- An unknown error HOT 32
- [BUG] summary and log are confusing. HOT 6
- "Number of mapped reads" from log file HOT 3
- [Feature Request] report number of duplicated fragments in bulk HOT 4
- Different ValidPairs rate between chromap and bowtie2 in HiC data HOT 9
- how to keep multi-mapped paires for HiC data. HOT 1
- [BUG] output to /dev/stdout HOT 6
- Understanding the multi-mapping reads and whether they are part of the bed file HOT 2
- ATAC-seq single end? HOT 3
- Coordinate system of the output fragment file? HOT 1
- multi-mapped reads HOT 3
- [BUG] Manpage is down HOT 1
- [BUG] Support for combinatorial barcode indexing(like SHARE) not present HOT 3
- [BUG] chromap map Hi-C short reads Parameters: error threshold HOT 2
- [BUG]For HiC data, the size of SAM files outputted using Chromap is much smaller compared to those from BWA-MEM HOT 4
- Repetitive or low-quality barcode sequences in scATAC data HOT 1
- [BUG] possibly improper MD tag generation whej running atac data. HOT 3
- Mapping paired-end single-cell ATAC-Seq reads HOT 2
- why so slow? HOT 2
- Failure to load cellular barcodes containing Ns HOT 3
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.
from chromap.