
Robust comparative analysis and contamination removal for metagenomics
![]()








With Recentrifuge, researchers can interactively explore what organisms are in their samples and at which level of confidence, thus enabling a robust comparative analysis of multiple samples in any metagenomic study.
Recentrifuge enables researchers to analyze results from Centrifuge, LMAT, CLARK, Kraken, and many other taxonomic classifiers using interactive pie charts, by placing great emphasis on the confidence level (score) of the taxonomic classifications.
The arithmetic of scored taxonomic trees of Recentrifuge supports the 48 taxonomic ranks of the NCBI Taxonomy, including several infraspecific levels such as strain or isolate. In addition to the scored charts for the original samples, different sets of scored plots are generated for each taxonomic level of interest, like shared taxa and exclusive taxa per sample.
If there are one or more negative control samples in the study, Recentrifuge will also generate additional control-subtracted interactive plots with the contamination removed from every sample and from the shared taxa specimen.
The novel and robust contamination removal algorithm of Recentrifuge detects and selectively removes various types of pollutants, including crossovers.
Recentrifuge quickly analyzes complex metagenomic datasets using parallel computational algorithms. It is especially useful in the case of low microbial biomass studies and when a more reliable detection of minority organisms is needed, like in clinical, environmental, and forensic applications.
Beyond the standard confidence levels, Recentrifuge implements others devoted to variable length reads, very convenient for datasets generated by nanopore sequencers.
For usage and docs, please, see the Recentrifuge wiki or, depending on your classifier, installation and:
- Recentrifuge for Centrifuge users
- Recentrifuge for LMAT users
- Recentrifuge for CLARK users
- Recentrifuge for Kraken users
- Recentrifuge for users of other classifiers
To play with an example of webpage generated by Recentrifuge, click on the screenshot:
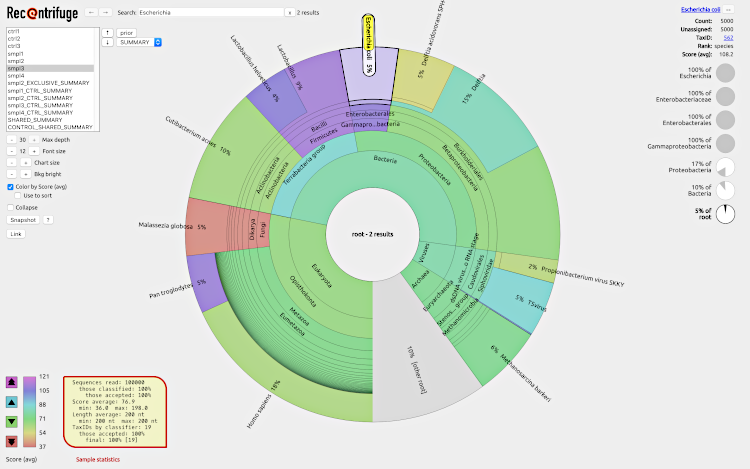
Recentrifuge is in a stable development status. For further details, please check the PLOS CB article.
If you use Recentrifuge in your research, please cite the paper. Thanks!
Martí JM (2019) Recentrifuge: Robust comparative analysis and contamination removal for metagenomics. PLOS Computational Biology 15(4): e1006967. https://doi.org/10.1371/journal.pcbi.1006967




























































































