marionilab / dropletutils Goto Github PK
View Code? Open in Web Editor NEWClone of the Bioconductor repository for the DropletUtils package.
Home Page: https://bioconductor.org/packages/devel/bioc/html/DropletUtils.html
Clone of the Bioconductor repository for the DropletUtils package.
Home Page: https://bioconductor.org/packages/devel/bioc/html/DropletUtils.html












































































I had previously asked about this problem last November, and was recommended to try the writefix branch, which I cant seem to find (not referenced in issue #31).
I am attempting to write a sparse matrix to h5 file using write10xcounts, the matrix was generated using the Read10X function from Seurat. Write10xcounts itself runs fine, however when running cellranger aggr on the resulting .h5 files, the following error is encountered, as before:
[error] The molecule info HDF5 file (write10xcountstest.h5) was produced by an older version of Cell Ranger. Reading these files is unsupported.
I replicated the example given at the bottom of the page here:
https://rdrr.io/github/MarioniLab/DropletUtils/man/write10xCounts.html
which produced the same error, therefore issue #31 has not been fixed.
sessionInfo below:
sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.6 LTS
Matrix products: default
BLAS: /opt/R-3.6.1/lib/libRblas.so
LAPACK: /opt/R-3.6.1/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8 LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
[7] LC_PAPER=en_GB.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] Matrix_1.2-18 DropletUtils_1.7.7 githubinstall_0.2.2 SingleCellExperiment_1.6.0 SummarizedExperiment_1.14.1
[6] DelayedArray_0.10.0 BiocParallel_1.18.1 matrixStats_0.55.0 Biobase_2.44.0 GenomicRanges_1.36.1
[11] GenomeInfoDb_1.20.0 IRanges_2.18.2 S4Vectors_0.22.1 BiocGenerics_0.30.0 cowplot_1.0.0
[16] ggplot2_3.2.1 dplyr_0.8.3 Seurat_3.1.1.9016
loaded via a namespace (and not attached):
[1] backports_1.1.4 plyr_1.8.4 igraph_1.2.4.1 lazyeval_0.2.2 splines_3.6.1 listenv_0.7.0 usethis_1.5.1
[8] digest_0.6.20 htmltools_0.4.0 gdata_2.18.0 magrittr_1.5 memoise_1.1.0 cluster_2.1.0 ROCR_1.0-7
[15] limma_3.40.6 remotes_2.1.0 globals_0.12.4 RcppParallel_4.4.4 R.utils_2.9.0 prettyunits_1.0.2 colorspace_1.4-1
[22] ggrepel_0.8.1 callr_3.3.1 crayon_1.3.4 RCurl_1.95-4.12 jsonlite_1.6 zeallot_0.1.0 survival_2.44-1.1
[29] zoo_1.8-6 ape_5.3 glue_1.3.1 gtable_0.3.0 zlibbioc_1.30.0 XVector_0.24.0 leiden_0.3.1
[36] pkgbuild_1.0.5 Rhdf5lib_1.6.3 future.apply_1.3.0 HDF5Array_1.12.3 scales_1.0.0 edgeR_3.26.8 bibtex_0.4.2
[43] Rcpp_1.0.2 metap_1.1 viridisLite_0.3.0 reticulate_1.13 dqrng_0.2.1 rsvd_1.0.2 SDMTools_1.1-221.1
[50] tsne_0.1-3 htmlwidgets_1.3 httr_1.4.1 gplots_3.0.1.1 RColorBrewer_1.1-2 ellipsis_0.3.0 ica_1.0-2
[57] pkgconfig_2.0.2 R.methodsS3_1.7.1 uwot_0.1.4 locfit_1.5-9.1 tidyselect_0.2.5 rlang_0.4.0 reshape2_1.4.3
[64] munsell_0.5.0 tools_3.6.1 cli_1.1.0 devtools_2.2.1 ggridges_0.5.1 stringr_1.4.0 npsurv_0.4-0
[71] processx_3.4.1 fs_1.3.1 fitdistrplus_1.0-14 caTools_1.17.1.2 purrr_0.3.2 RANN_2.6.1 pbapply_1.4-2
[78] future_1.14.0 nlme_3.1-140 R.oo_1.22.0 compiler_3.6.1 rstudioapi_0.10 plotly_4.9.0 curl_4.0
[85] png_0.1-7 testthat_2.2.1 lsei_1.2-0 tibble_2.1.3 stringi_1.4.3 ps_1.3.0 desc_1.2.0
[92] lattice_0.20-38 vctrs_0.2.0 pillar_1.4.2 lifecycle_0.1.0 Rdpack_0.11-0 lmtest_0.9-37 RcppAnnoy_0.0.13
[99] data.table_1.12.8 bitops_1.0-6 irlba_2.3.3 gbRd_0.4-11 R6_2.4.0 KernSmooth_2.23-15 gridExtra_2.3
[106] sessioninfo_1.1.1 codetools_0.2-16 MASS_7.3-51.4 gtools_3.8.1 assertthat_0.2.1 pkgload_1.0.2 rhdf5_2.28.1
[113] rprojroot_1.3-2 withr_2.1.2 sctransform_0.2.0 GenomeInfoDbData_1.2.1 grid_3.6.1 tidyr_1.0.0 Rtsne_0.15
I am attempting to reproduce your example for swappedDrops() with HiSeq4000 data from the report bcswap_supp.Rmd (in repo BarcodeSwapping2017), but this step gets stuck in an endless loop:
cleaned_4000 <- swappedDrops(h5_files, get.diagnostics = TRUE, get.swapped = TRUE)
My R session info is as follows:
R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] igraph_1.2.4 DropletUtils_1.2.2 Rtsne_0.15 scater_1.10.1 scran_1.10.2 SingleCellExperiment_1.4.1 SummarizedExperiment_1.12.0 DelayedArray_0.8.0 matrixStats_0.54.0
[10] Biobase_2.42.0 GenomicRanges_1.34.0 GenomeInfoDb_1.18.2 IRanges_2.16.0 S4Vectors_0.20.1 BiocGenerics_0.28.0 BiocParallel_1.16.6 irlba_2.3.3 limSolve_1.5.5.3
[19] scales_1.0.0 Matrix_1.2-14 BiocStyle_2.10.0 knitr_1.22 here_0.1 RColorBrewer_1.1-2 viridis_0.5.1 viridisLite_0.3.0 cowplot_0.9.4
[28] reshape2_1.4.3 ggplot2_3.1.1
loaded via a namespace (and not attached):
[1] dynamicTreeCut_1.63-1 edgeR_3.24.3 DelayedMatrixStats_1.4.0 assertthat_0.2.1 statmod_1.4.30 BiocManager_1.30.4 vipor_0.4.5 GenomeInfoDbData_1.2.0 yaml_2.2.0 pillar_1.3.1
[11] backports_1.1.4 lattice_0.20-35 glue_1.3.1 limma_3.38.3 quadprog_1.5-5 digest_0.6.18 XVector_0.22.0 colorspace_1.4-1 htmltools_0.3.6 plyr_1.8.4
[21] lpSolve_5.6.13 pkgconfig_2.0.2 zlibbioc_1.28.0 purrr_0.3.2 HDF5Array_1.10.1 tibble_2.1.1 withr_2.1.2 lazyeval_0.2.2 magrittr_1.5 crayon_1.3.4
[31] evaluate_0.13 MASS_7.3-50 beeswarm_0.2.3 tools_3.5.1 stringr_1.4.0 Rhdf5lib_1.4.3 munsell_0.5.0 locfit_1.5-9.1 compiler_3.5.1 rlang_0.3.4
[41] rhdf5_2.26.2 grid_3.5.1 RCurl_1.95-4.12 BiocNeighbors_1.0.0 bitops_1.0-6 rmarkdown_1.12 gtable_0.3.0 R6_2.4.0 gridExtra_2.3 dplyr_0.8.0.1
[51] rprojroot_1.3-2 stringi_1.4.3 ggbeeswarm_0.6.0 Rcpp_1.0.1 tidyselect_0.2.5 xfun_0.6
Thank you in advance!
Would this be difficult?
I've got some samples processed with CellRanger v3 (barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz), which the version of read10xCounts() in the release branch can't handle.
Unfortunately, I can't switch to devel because IT reasons.
For now, I've just worked around it by copying the relevant code to a local script, but it'd be cool to have since there's a few months yet until devel becomes release.
Hi Aaron,
I am able to run the function read10xMolInfo without error, but when I run downsampleReads I get the following: "Error: segfault from C stack overflow". I have R version 3.6.1 and DropletUtils_1.4.2. The 10x molecule.info file is from a 10x 3' v3 kit, which read10xMolInfo is able to detect. Can you offer any advice? The commands I used and the sessionInfo() are below.
Thanks!
readmolinfo <- read10xMolInfo(moleculeinfo_path, barcode.length=16)
nodownsample <- downsampleReads(moleculeinfo_path, prop = 1, barcode.length=16, bycol=FALSE)
Error: segfault from C stack overflow
sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: Ubuntu 16.04.4 LTS
Matrix products: default
BLAS/LAPACK: /data/yosef2/users/zsteier/programs/miniconda/envs/Renv3_6/lib/libopenblasp-r0.3.6.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DropletUtils_1.4.2 SingleCellExperiment_1.6.0
[3] SummarizedExperiment_1.14.1 DelayedArray_0.10.0
[5] BiocParallel_1.18.0 matrixStats_0.54.0
[7] Biobase_2.44.0 GenomicRanges_1.36.0
[9] GenomeInfoDb_1.20.0 IRanges_2.18.1
[11] S4Vectors_0.22.0 BiocGenerics_0.30.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.2 edgeR_3.26.6 XVector_0.24.0
[4] zlibbioc_1.30.0 lattice_0.20-38 tools_3.6.1
[7] grid_3.6.1 rhdf5_2.28.0 dqrng_0.2.1
[10] R.oo_1.22.0 HDF5Array_1.12.1 Matrix_1.2-17
[13] GenomeInfoDbData_1.2.1 Rhdf5lib_1.6.0 R.utils_2.9.0
[16] bitops_1.0-6 RCurl_1.95-4.12 limma_3.40.6
[19] compiler_3.6.1 R.methodsS3_1.7.1 locfit_1.5-9.1
Hi,
I am a noive in DropletUtils, I used read10xMolInfo() to load a HDF5 file. It works well. But when I run the downsampleReads() for the mol.info.file, I got error
Error: Error in h5checktypeOrOpenLoc(). Argument neither of class H5IdComponent nor a character.
Can you help me fix it?
Thanks!
Error: object ‘whichNonZero’ is not exported by 'namespace:beachmat'
Execution halted
ERROR: lazy loading failed for package ‘DropletUtils’
* removing ‘/Library/Frameworks/R.framework/Versions/4.0/Resources/library/DropletUtils’
Warning message:
In i.p(...) :
installation of package ‘/var/folders/vr/m2d68y7d3xs36599czqs6fwc0000gn/T//RtmpCU2qfE/filef8b2419935e6/DropletUtils_1.11.12.tar.gz’ had non-zero exit status
Release 3_12 installed without a problem.
R version 4.0.4 (2021-02-15)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] compiler_4.0.4
Hi,
I wonder whether it is necessary to run emptydrops function for count matrix obtained from 10x cell ranger 3.0 and above? The following description for cell ranger indicates that the algorithm is already using emptydrops internally?
'Cell Ranger 3.0 introduces an improved cell-calling algorithm that is better able to identify populations of low RNA content cells, especially when low RNA content cells are mixed into a population of high RNA content cells. For example, tumor samples often contain large tumor cells mixed with smaller tumor infiltrating lymphocytes (TIL) and researchers may be particularly interested in the TIL population. The new algorithm is based on the EmptyDrops method (Lun et al., 2018).'
Hi, I encounter an error that is similar to the one I reported in scater's repo when I ran emptyDrops.
Error in .local(x, ...): cluster already started
Traceback:
1. emptyDrops(mat, BPPARAM = bpp)
2. emptyDrops(mat, BPPARAM = bpp)
3. .local(m, ...)
4. testEmptyDrops(m, lower = lower, round = FALSE, test.ambient = test.ambient,
. ..., BPPARAM = BPPARAM)
5. bpstart(BPPARAM)
6. bpstart(BPPARAM)
7. .local(x, ...)
8. stop("cluster already started")
Error in serialize(data, node$con, xdr = FALSE): ignoring SIGPIPE signal
Traceback:
1. emptyDrops(mat, BPPARAM = bpp)
2. emptyDrops(mat, BPPARAM = bpp)
3. .local(m, ...)
4. bpstop(BPPARAM)
5. bpstop(BPPARAM)
6. .bpstop_impl(x)
7. .bpstop_nodes(x)
8. .send_to(cluster, i, .DONE())
9. .send_to(cluster, i, .DONE())
10. parallel:::sendData(backend[[node]], value)
11. sendData.SOCK0node(backend[[node]], value)
12. serialize(data, node$con, xdr = FALSE)
I noticed in the source codes
Line 190 in 71296b6
and
Line 370 in 71296b6
probably should be like below as LTLA pointed out in my original issue in scater
if (.bpNotSharedOrUp(BPPARAM)) {
Can't run downsamplereads or read10xMolInfo on my h5 file. I have tried using different versions of DropletUtils (including most up to date) in in both R 4.0.2 and R 3.6.1. In most cases (besides v1.9) can get example script to run. See my script below. Any help would be appreciated. I attached error code for this particular trial but sometimes running the same exact script I get the error.
"Error in read10xMolInfo(sample, barcode.length, get.umi = FALSE) :
c++ exception (unknown reason)"
10x info is as follows Chemistry | Single Cell 3' v3 Pipeline Version | 3.1.0
######################Script############################
library(DropletUtils)
#DropletUtils v1.6.1
#R version 4.0.2 (2020-06-22)
out <- "Z:\Bioinformatics\Students_Fellows\Snowball\pctc\Normal_Human_Epi_Mes\Cellranger\hg38\filtered_feature_bc_matrix.h5"
read10xMolInfo(out)
hg38_downsample <- downsampleReads(out, prop=.2, barcode.length=NULL, bycol=FALSE)
#############Example Script that works############
out <- DropletUtils:::sim10xMolInfo(tempfile(), nsamples=1)
out2 <- downsampleReads(out, barcode.length=0, prop=0.5)
##############Error######################
hg38_downsample <- downsampleReads(out, prop=.2, barcode.length=NULL, bycol=FALSE)
: Symbol table
minor: Callback failed
#5: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
HDF5-DIAG: Error detected in HDF5 (1.10.6) thread 0:
#000: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5D.c line 298 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#1: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Dint.c line 1410 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#2: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#3: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 851 in H5G_traverse(): internal path traversal failed
major: Symbol table
minor: Object not found
#4: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 627 in H5G__traverse_real(): traversal operator failed
major: Symbol table
minor: Callback failed
#5: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
HDF5-DIAG: Error detected in HDF5 (1.10.6) thread 0:
#000: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5D.c line 298 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#1: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Dint.c line 1410 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#2: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#3: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 851 in H5G_traverse(): internal path traversal failed
major: Symbol table
minor: Object not found
#4: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 627 in H5G__traverse_real(): traversal operator failed
major: Symbol table
minor: Callback failed
#5: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
HDF5-DIAG: Error detected in HDF5 (1.10.6) thread 0:
#000: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5D.c line 298 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#1: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Dint.c line 1410 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#2: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#3: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 851 in H5G_traverse(): internal path traversal failed
major: Symbol table
minor: Object not found
#4: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 627 in H5G__traverse_real(): traversal operator failed
major: Symbol table
minor: Callback failed
#5: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
HDF5-DIAG: Error detected in HDF5 (1.10.6) thread 0:
#000: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5D.c line 298 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#1: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Dint.c line 1410 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#2: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#3: C:/rtools40/tmp/CMake-hdf5-1.10.6/hdf5-1.10.6/src/H5Gtraverse.c line 851 in H5G_traverse(): Error in read10xMolInfo(sample, barcode.length, get.umi = FALSE) :
c++ exception (unknown reason)
hg38_downsample <- downsampleReads(out, prop=.2, barcode.length=NULL, bycol=FALSE)
Error in read10xMolInfo(sample, barcode.length, get.umi = FALSE) :
c++ exception (unknown reason)
Hi, I'm using the BD Rhapsody scRNA technique. We sequenced 100G data for one sample and obtained maybe 9000 nuclei (not cell).
I'm using the only output matrix which I assume is the raw (I'm not 100% sure [upate: this is NOT correct, see my next comment]). The knee and inflection are 326 and 144, respectively (following this tutorial https://bioconductor.org/packages/release/bioc/vignettes/DropletUtils/inst/doc/DropletUtils.html)
When using the default value in emptyDrops (lower = 100), no output obtained as mentioned here (#29). Apparently, because no ambient exists ( colSum < 100).
I change the "lower" to 150 or 200 which works well. But the results are different.
For example, when using 150, the cell number is 8670, and empty is 1187; when using 200, the cell number is 7232, empty 2197 and NA 436.
I understand that "lower" is the total umi per nuclei in my case, but not sure how can I decide this parameter since it highly affects the result?
Thank you very much!

Originally posted by @hk20013106 in #29 (comment)
Often, data obtained from GEO Browser will be a TAR file of all of the samples' files. For example,
$ ls | head
GSE139324_RAW.tar
GSM4138110_HNSCC_1_PBMC_barcodes.tsv.gz
GSM4138110_HNSCC_1_PBMC_genes.tsv.gz
GSM4138110_HNSCC_1_PBMC_matrix.mtx.gz
GSM4138111_HNSCC_1_TIL_barcodes.tsv.gz
GSM4138111_HNSCC_1_TIL_genes.tsv.gz
GSM4138111_HNSCC_1_TIL_matrix.mtx.gz
GSM4138112_HNSCC_2_PBMC_barcodes.tsv.gz
GSM4138112_HNSCC_2_PBMC_genes.tsv.gz
GSM4138112_HNSCC_2_PBMC_matrix.mtx.gz
But from the description of samples, read10xCounts requires that each sample be in a separate folder. Could it be modified so that it works easily with data obtained from GEO Browser?
Hello Aaron,
I used function swappedDrops() to do preprocessing, but when I ran codes as follow:
library(ggplot2)
library(cowplot)
library(Matrix)
mol_loc = "./Desktop/Single_cell/cJUN/Data/119-13cJUN-kowt/filtered_feature_bc_matrix.h5"
out_loc = "./Desktop/Single_cell/cJUN/Data/119-13cJUN-kowt/filtered_feature_bc_matrix/matrix.mtx.gz"
bc_loc = "./Desktop/Single_cell/cJUN/Data/119-13cJUN-kowt/filtered_feature_bc_matrix/barcodes.tsv.gz"
gene_loc = "./Desktop/Single_cell/cJUN/Data/119-13cJUN-kowt/filtered_feature_bc_matrix/features.tsv.gz"
unswapped = swappedDrops(mol_loc,get.swapped = T)
I got the error like this:
> unswapped = swappedDrops(mol_loc,get.swapped = T)
HDF5-DIAG: Error detected in HDF5 (1.10.5) thread 0:
#000: H5D.c line 292 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#001: H5Dint.c line 1384 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#002: H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#003: H5Gtraverse.c line 851 in H5G_traverse(): internal path traversal failed
major: Symbol table
minor: Object not found
#004: H5Gtraverse.c line 627 in H5G__traverse_real(): traversal operator failed
major: Symbol table
minor: Callback failed
#005: H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
Error in read10xMolInfo(samples[i], barcode.length = barcode.length) :
c++ exception (unknown reason)
I had looked at #10, and tried methods you mentioned in that issue. It still gave the same error.
I obtained dataset by running CellRanger v3.0.2.
Session information:
> sessionInfo()
R version 3.6.0 (2019-04-26)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] Matrix_1.2-18 cowplot_1.0.0 ggplot2_3.2.1
[4] DropletUtils_1.6.1 SingleCellExperiment_1.8.0 SummarizedExperiment_1.16.1
[7] DelayedArray_0.12.2 BiocParallel_1.20.1 matrixStats_0.55.0
[10] Biobase_2.46.0 GenomicRanges_1.38.0 GenomeInfoDb_1.22.0
[13] IRanges_2.20.2 S4Vectors_0.24.2 BiocGenerics_0.32.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.3 locfit_1.5-9.1 lattice_0.20-38 prettyunits_1.1.0
[5] ps_1.3.0 rprojroot_1.3-2 assertthat_0.2.1 digest_0.6.23
[9] R6_2.4.1 backports_1.1.5 pillar_1.4.3 zlibbioc_1.32.0
[13] rlang_0.4.2 curl_4.3 lazyeval_0.2.2 rstudioapi_0.10
[17] callr_3.4.0 R.utils_2.9.2 R.oo_1.23.0 desc_1.2.0
[21] devtools_2.2.1 RCurl_1.95-4.12 munsell_0.5.0 HDF5Array_1.14.1
[25] compiler_3.6.0 pkgconfig_2.0.3 pkgbuild_1.0.6 tidyselect_0.2.5
[29] tibble_2.1.3 GenomeInfoDbData_1.2.2 edgeR_3.28.0 fansi_0.4.1
[33] crayon_1.3.4 dplyr_0.8.3 withr_2.1.2 bitops_1.0-6
[37] R.methodsS3_1.7.1 grid_3.6.0 gtable_0.3.0 lifecycle_0.1.0
[41] magrittr_1.5 scales_1.1.0 dqrng_0.2.1 cli_2.0.1
[45] XVector_0.26.0 fs_1.3.1 remotes_2.1.0 testthat_2.3.1
[49] limma_3.42.0 ellipsis_0.3.0 Rhdf5lib_1.8.0 tools_3.6.0
[53] glue_1.3.1 purrr_0.3.3 pkgload_1.0.2 processx_3.4.1
[57] yaml_2.2.0 colorspace_1.4-1 rhdf5_2.30.1 sessioninfo_1.1.1
[61] memoise_1.1.0 usethis_1.5.1
Hello,
I was trying to identify droplets on filtered data in which only barcodes with at least 100 nUMI were kept. The emptyDrops got stuck and never ended. I am curious if the input must be original count matrix from 10X rather than filtered one. Thanks for your help.
Best,
Yang
Hello,
I've recently tried to "replicate" cellranger analysis by running STAR solo and then empty drops on the unfiltered data. STAR solo gives me the correct (as in, the same as cellranger 3.0) number of unfiltered droplets, but emptyDrops is not so faithful, as i mentioned here: alexdobin/STAR#879
I am getting nearly 6x the count that I got with cellranger 3. Do you have any insight as to what parameters I can tweak in emptyDrops to get the same count? It would save us some time in testing if this was already a solved problem.
I appreciate your time in advance. Have a great weekend!
Hi Aaron,
I used the DropletUtils to filter data.
I countered this problem .
tmpdir = setwd("F:/sample208_filtered_feature_bc_matrix") dir.name <- tmpdir list.files(dir.name)
sce <- read10xCounts(dir.name,col.names = TRUE) sce
dim: 32738 11701
metadata(0):
assays(1): counts
rownames(32738): ENSG00000243485 ENSG00000237613 ... ENSG00000215616
ENSG00000215611
rowData names(3): ID Symbol NA
colnames(11701): AAACCCAAGACAAGCC-1 AAACCCAAGCTGTACT-1 ... TTTGTTGTCGATACGT-1
TTTGTTGTCTCTGACC-1
colData names(2): Sample Barcode
reducedDimNames(0):
spikeNames(0):
set.seed(100) e.out <- emptyDrops(counts(sce))
Error in testEmptyDrops(m, lower = lower, ...) : no counts available to estimate the ambient profile
I don't know this problem about relativing "rowData names(3):ID Symbol NA"
I will appreciate any advice on how to solve this.
Many thanks
Janexty
Hi,
Updating to 1.5.10 run into following errors;
clang-9: error: no such file or directory: '"/Users/ahoji/Documents/R_lib/Rhdf5lib/lib/libhdf5_cpp.a"'
clang-9: error: no such file or directory: '"/Users/ah/Documents/R_lib/Rhdf5lib/lib/libhdf5.a"'
clang-9: error: no such file or directory: '"/Users/ah/Documents/R_lib/Rhdf5lib/lib/libsz.a"'
even though those 3 files are there;
(base) ➜ lib ls libhdf5.a libhdf5_cpp.a libhdf5_hl.a libhdf5_hl_cpp.a libsz.a
Any help will be appreciated
Hello,
Is there a way to specify different proportions for different rows in downsampleReads? We have data that contains gene expression plus feature barcoding (generated by cellranger count 3.0) but some of the feature barcoding runs were from different sequencing runs. So we don't necessarily want to specify the same proportion for the gene and protein features.
I know we can also do downsampleMatrix and handle the feature barcoding and gene expression matrices separately, but because the documentation notes "Technically, downsampling on the reads with downsampleReads is more appropriate as it recapitulates the effect of differences in sequencing depth per cell" we thought we'd also check out downsampleReads.
Thank you! I apologize if I am overlooking something but I did not see this in the documentation.
Hi Aaron,
In the documentation of the barcodeRanks, it said that the input matrix, m, should have "Columns represent barcoded droplets, rows represent cells".
Is this a typo that the columns should be "genes" and rows are cells (or the other way around) Or have I misunderstood this function and it expects a cell-filtered data as the input?
Many thanks,
Kevin
Hello,
I just upgraded to Bioconductor version 3.9.
When I run emptyDrops() on my count data it does not terminate (i.e. endless loop? goes on for hours). I have tried changing niters to be lower (i.e 100) but nothing seems to work.
When I revert back to Bioconductor version 3.8, emptyDrops works and terminates fine.
There are few emptyDrops detected overall (i.e. 5) but I would still like to know why it doesn't terminate when using the latest version. My count data is ~33K genes x ~1500 cells
Thanks
shui
The speed of running both emptyDrops and estimateAmbience is very slow if the count matrix input is a DelayedMatrix or TENxMatrix object.
I understand the documentation did mentioned the expected input is either a dgTMatrix or dgCMatrix. However, as read10xCounts also works with a HDF5 input and stores counts as DelayedMatrix, it will be great if the functions are also DelayedArray-friendly, maybe a check on the object class and convert to dgCMatrix if necessary at the beginning of the operation?
Hello MarioniLab!
We've seen some pretty extreme ambient RNA effects in our data, but when I enter my dgCMatrix, I get this error:
testEmptyDrops(fivemonth.data, lower=100, niters=10000, test.ambient=FALSE, ignore=NULL, alpha=NULL, BPPARAM=SerialParam())
"Error in testEmptyDrops(fivemonth.data, lower = 100, niters = 10000, test.ambient = FALSE, :
no counts available to estimate the ambient profile"
I'm running on windows in RStudio with R 3.6.1.
Hope you can help and thanks for reading this!
Casey
Hi,
I have tried to install the new version of the package using:
devtools::install_github("MarioniLab/DropletUtils")
and got the following error:
ERROR: compilation failed for package ‘DropletUtils’
Thanks
This was brought up by @LTLA in issue #33 as possible ways to avoid issues when emptyDrops fails to properly identify the knee point. Here is my experience with a particular sample:
Barcode ranks plot fails to correctly find knee.
bcrank <- barcodeRanks(counts)
# Only showing unique points for plotting speed.
uniq <- !duplicated(bcrank$rank)
plot(bcrank$rank[uniq], bcrank$total[uniq], log="xy",
xlab="Rank", ylab="Total UMI count", cex.lab=1.2)
abline(h=metadata(bcrank)$inflection, col="darkgreen", lty=2)
abline(h=metadata(bcrank)$knee, col="dodgerblue", lty=2)
legend("bottomleft", legend=c("Inflection", "Knee"),
col=c("darkgreen", "dodgerblue"), lty=2, cex=1.2)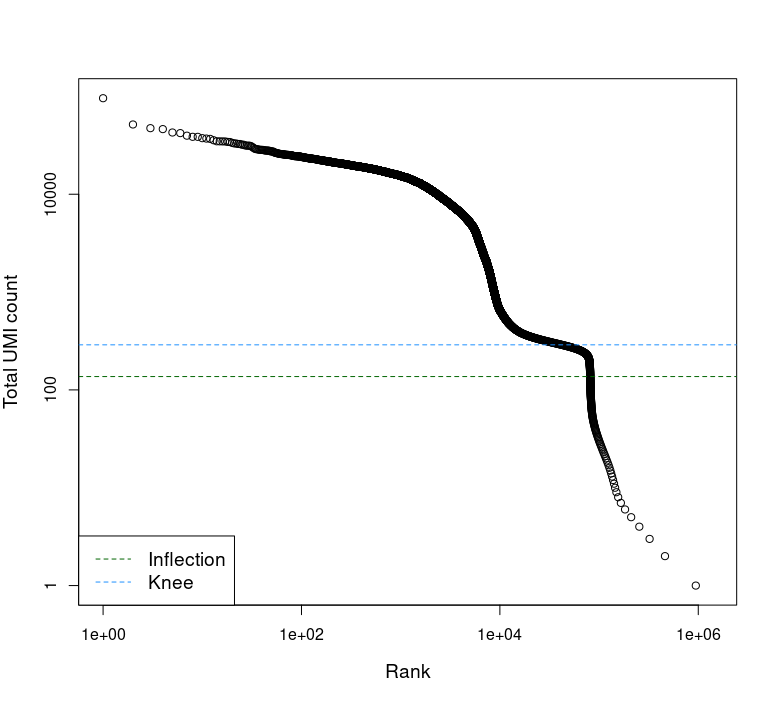
Default call to empty drops unsurprisingly retains too many cells (everything above the knee will be retained).
# default parameters
set.seed(100)
e.out <- DropletUtils::emptyDrops(counts)
summary(e.out$FDR < 0.001)
# Mode FALSE TRUE NA's
# logical 26965 55104 1358742 Setting retain = Inf so that knee isn't used:
set.seed(100)
e.out <- DropletUtils::emptyDrops(counts, retain = Inf)
summary(e.out$FDR < 0.001)
# Mode FALSE TRUE NA's
# logical 54136 27933 1358742 Removing mitochondrial and ribosomal genes so that they don't get used when determining if droplets are different from ambient droplets:
# qc is a list of character vectors with mitochondrial and ribosomal gene names
keep <- !row.names(counts) %in% unlist(qc)
set.seed(100)
e.out <- DropletUtils::emptyDrops(counts[keep, ])
summary(e.out$FDR < 0.001)
# Mode FALSE TRUE NA's
# logical 73095 8411 1359305
# for completeness, also with retain = Inf
set.seed(100)
e.out <- DropletUtils::emptyDrops(counts[keep, ], retain = Inf)
summary(e.out$FDR < 0.001)
# Mode FALSE TRUE NA's
#logical 73095 8411 1359305 Hi:
I can't seem to get swappedDrops to run due to what appears to be some sort of file handling issue(?). Everything worked flawlessly when I followed the instructions to mock up the data and run swappedDrops, but every time I try to use "real" molecule_info.h5 files from 10X, I get the "Missing filters: deflate" error detailed below. I've tried using in-house generated molecule_info.h5 files (processed via Cell Ranger 3.1.0) as well as ones downloaded from 10X (also from Cell Ranger 3.1.0).
I noticed that a previously reported issue described a similar problem, but it sounded like the problem in that case was an incompatibility between an earlier version of DropletUtils (version 1.2.2) and Cell Ranger 3.1. The solution there appeared to be an update to DropletUtils version 1.3.10, but I'm assuming that the current version of DropletUtils (1.4.3) is the correct version to use?
Apologies in advance if I'm just being dense and missing something obvious. And many, many thanks in advance for any help/guidance you can offer.
Session output:
suppressMessages(library(DropletUtils))
Warning messages:
1: package ‘SingleCellExperiment’ was built under R version 3.6.1
2: package ‘BiocGenerics’ was built under R version 3.6.1
3: package ‘GenomeInfoDb’ was built under R version 3.6.1
4: package ‘Biobase’ was built under R version 3.6.1
5: package ‘DelayedArray’ was built under R version 3.6.1
6: package ‘matrixStats’ was built under R version 3.6.1
mol.info <- c("/home/ubuntu/test_swapped_drops/h5dir/135_molecule_info.h5","/home/ubuntu/test_swapped_drops/h5dir/136_molecule_info.h5")
mol.info
[1] "/home/ubuntu/test_swapped_drops/h5dir/135_molecule_info.h5"
[2] "/home/ubuntu/test_swapped_drops/h5dir/136_molecule_info.h5"
out <- swappedDrops(mol.info)
Error: Unable to read dataset.
Not all required filters available.
Missing filters: deflate
sessionInfo()
R version 3.6.0 (2019-04-26)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: Ubuntu 18.04.2 LTS
Matrix products: default
BLAS/LAPACK: /home/ubuntu/miniconda3/lib/R/lib/libRblas.so
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] DropletUtils_1.4.3 SingleCellExperiment_1.6.0
[3] SummarizedExperiment_1.14.1 DelayedArray_0.10.0
[5] BiocParallel_1.18.1 matrixStats_0.55.0
[7] Biobase_2.44.0 GenomicRanges_1.36.1
[9] GenomeInfoDb_1.20.0 IRanges_2.18.3
[11] S4Vectors_0.22.1 BiocGenerics_0.30.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.2 edgeR_3.26.8 XVector_0.24.0
[4] zlibbioc_1.30.0 lattice_0.20-38 tools_3.6.0
[7] grid_3.6.0 rhdf5_2.28.1 dqrng_0.2.1
[10] R.oo_1.23.0 HDF5Array_1.12.3 Matrix_1.2-17
[13] GenomeInfoDbData_1.2.2 Rhdf5lib_1.6.3 R.utils_2.9.0
[16] bitops_1.0-6 RCurl_1.95-4.12 limma_3.40.6
[19] compiler_3.6.0 R.methodsS3_1.7.1 locfit_1.5-9.1
Hi Aaron,
Great package!
I am a bit of a newbie to R. I am interested in looking at UMI duplication, how would i go about to export the read10xMolInfo output into a .csv and decoding the integer 2bit encoded values into ATCG? I basically want a .csv with a column of barcode, umi, count, GeneName, EnselmbeID.
Thank you! Oh also, thank you for all the tutorials, pretty much half the people i know have used them to learn how-to scRNA :)
Hi,
I am looking into the details of EmptyDrops and I have a bit of a hard time understanding a detail regarding the approach used.
In the article, you state that using the Good-Turing algorithm "ensures that genes with zero counts in the ambient pool have non-zero proportions, avoiding undefined likelihoods in downstream calculations. " and that the likelihood equation "highlights the usefulness of pg instead of a naive estimate of the proportions from A, as the former is guaranteed to be non-zero such that Lb is defined."
However, looking into the Good-Turing algorithm, it is clear that the estimated posterior for a gene with ambient expression 0 is non-zero if and only if there is at least one gene with ambient expression 1. I have verified this by using goodTuringProportions from edgeR.
So the first question is, how likely is it for that to happen and end up with undefined likelihood?
The second question is, are you dealing with this situation separately in the code? I have generated data that should lead to undefined likelihoods, however the emptyDrops function seems to work just fine in this situation. Here is the code I used.
noGenes <- 100
noBarcodes <- 1000
expressionMatrix <- sapply(1:noBarcodes, function(i) {rnbinom(noGenes, mu = 100, size = 1/50)})
totalExpression <- colSums(expressionMatrix)
summary(totalExpression)
lower <- round(quantile(totalExpression, probs = 0.25))
ambientBarcodes <- which(totalExpression <= lower)
remainingBarcodes <- setdiff(1:noBarcodes, ambientBarcodes)
length(ambientBarcodes)
# Add genes with expression 0 in the ambient profile
noSpecialGenes <- 10
specialGenes <- matrix(0, ncol = noBarcodes, nrow = noSpecialGenes)
specialGenes[, remainingBarcodes] <- sapply(1:length(remainingBarcodes), function(i) {rnbinom(noSpecialGenes, mu = 100, size = 1/50)})
summary(rowSums(specialGenes))
expressionMatrix <- rbind(expressionMatrix, specialGenes)
ambientProfile <- rowSums(expressionMatrix[, ambientBarcodes])
summary(ambientProfile)
posterior <- goodTuringProportions(ambientProfile)
summary(posterior)
# Remove any gene with expression 1 in the ambient profile
pos <- which(ambientProfile == 1)
summary(pos)
if (length(pos) > 0) {
expressionMatrix <- expressionMatrix[-pos, ]
}
testing <- emptyDrops(expressionMatrix, lower = lower)
summary(testing$PValue)
Thank you!
Paula
Hello, I noticed the Bioconductor release for R3.6 (Bioc 3.10) only has DropletUtils 1.6.1 is there any plan to update Bioc 3.10 to the newer version? Or does the latest version have some R4.x specific code?
Hi,
Looking at the documentation for the emptyDrops function, I am confused as to whether it is necessary to perform "testEmptyDrops" prior to running "emptyDrops"? From the example, it doesn't seem as though this is necessary but then the information says that emptyDrops uses the results from testEmptyDrops?
"The emptyDrops function combines the results of testEmptyDrops with barcodeRanks to identify droplets that are likely to contain cells"
I would appreciate your clarification on this.
Many thanks,
Lucy
Hi,
I am performing combined analysis of three scRNA-seq samples using Cell Ranger and Seurat. I am trying to understand the purpose of downsampling the matrix/reads with DropletUtils if you are then going to normalize for cell sequencing depth within the sctransform step in Seurat? Is there any advantage to performing both of these normalization steps or would you perform one or the other? In what situations would DropletUtils downsampling be advantageous over Seurat normalization and vice versa?
Many thanks,
Lucy
Hello,
I have some troubles to install DropletUtils, I followed the instruction here :
https://bioconductor.org/packages/devel/bioc/html/DropletUtils.html
But when I tried to update these old packages I have an error :
The downloaded source packages are in
‘/tmp/Rtmp2VmbCx/downloaded_packages’
Old packages: 'DelayedArray', 'GenomicFeatures', 'HDF5Array', 'limma'
Warning messages:
1: In install.packages(update[instlib == l, "Package"], l, contriburl = contriburl, :
installation of package ‘DelayedArray’ had non-zero exit status
2: In install.packages(update[instlib == l, "Package"], l, contriburl = contriburl, :
installation of package ‘GenomicFeatures’ had non-zero exit status
3: In install.packages(update[instlib == l, "Package"], l, contriburl = contriburl, :
installation of package ‘limma’ had non-zero exit status
4: In install.packages(update[instlib == l, "Package"], l, contriburl = contriburl, :
installation of package ‘HDF5Array’ had non-zero exit status
And when I installed them with install.packages() I get :
install.packages("DelayedArray")
Installing package into ‘/home/lea/R/x86_64-pc-linux-gnu-library/3.5’
(as ‘lib’ is unspecified)
Warning in install.packages :
package ‘DelayedArray’ is not available (for R version 3.5.1)
Here is my sessionInfo:
> sessionInfo()
R version 3.5.1 (2018-07-02)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 14.04.5 LTS
Matrix products: default
BLAS: /usr/lib/libblas/libblas.so.3.0
LAPACK: /usr/lib/lapack/liblapack.so.3.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=fr_FR.UTF-8 LC_COLLATE=en_US.UTF-8 LC_MONETARY=fr_FR.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=fr_FR.UTF-8
[8] LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] BiocInstaller_1.31.3
loaded via a namespace (and not attached):
[1] BiocManager_1.30.2 compiler_3.5.1 tools_3.5.1 yaml_2.2.0
Can you help me ?
Kind regards,
Lea
Hello,
I processed my scRNA-seq sample using cellranger 3.1.0. Now, I wanted to extract reads information each cellbarcode and the genes for which I used the rread10XMolInfo command to open the molecule_info.h5 file. But for some very strange reason as soon as I run this command in rstudio, it just doesn't load the files and it hangs the application. Meanwhile, when I used a molecule_infor.h5 file generated from another dataset that I processed using cellranger 2.1.1, it loads the file in a variable with no issues. I have been having a hard time figuring this out, so can you suggest a way to load the molecule_info.h5 file using Dropletutils and also to obtain a feature-bc matrix with read counts instead of UMIs?
Hi Aaron,
I've use DropletUtils to filtrate my single cell data coming from STARsolo.
I would like to proceed to Seurat, however I don't know how to obtain the filtrated matrix.mtx, barcodes.tvs and genes.tvs for this purpose.
I've loaded the unfiltered matrix.mtx, barcodes.tvs and genes.tvs, I've performed read10xCounts to pull this data together, then barcodeRanks and finally emptyDroplets. The final output I got is the is.cell logical as it's explained in Utilities for handling droplet-based single-cell RNA-seq data from Bioconductor.
is.cell <- e.out$FDR <= 0.01
sum(is.cell, na.rm=TRUE)
What am I missing?
Thank you for your time!
Best,
Lucia
It seems that write10xCounts's HDF5 output doesn't work when you put it back into CellRanger, due to the absence of some undocumented HDF5 attributes. This has now been fixed in the writefix branch though there may still be some residual problems from type differences (the integers are not quite stored as unsigned 32/64-bits and the strings are probably not stored as how CellRanger generates them; hopefully these are handled smoothly by PyTables).
my codes are under following:
library(githubinstall)
githubinstall("DropletUtils")
Suggestion:
how should I to re-try?
Hi Aaron,
I use the DropletUtils package to filter single cell data.
I have encountered this problem .
tmpdir = setwd("F:/sample208_filtered_feature_bc_matrix")
dir.name <- tmpdir
list.files(dir.name)
"barcodes.tsv" "genes.tsv" "matrix.mtx"
sce <- read10xCounts(dir.name,col.names = TRUE)
sce
class: SingleCellExperiment
dim: 32738 11701
metadata(0):
assays(1): counts
rownames(32738): ENSG00000243485 ENSG00000237613 ... ENSG00000215616
ENSG00000215611
rowData names(3): ID Symbol NA
colnames(11701): AAACCCAAGACAAGCC-1 AAACCCAAGCTGTACT-1 ... TTTGTTGTCGATACGT-1
TTTGTTGTCTCTGACC-1
colData names(2): Sample Barcode
reducedDimNames(0):
spikeNames(0):
set.seed(100)
e.out <- emptyDrops(counts(sce))
Error in testEmptyDrops(m, lower = lower, ...) : no counts available to estimate the ambient profile
I don't know this problem about relativing "rowData names(3):ID Symbol NA"
I will appreciate any advice on how to solve this.
Many thanks
Janexty
I get different results for the number of significant barcodes depending on how test.ambient is set. Here's an example:
fdr <- .001
lower <- 100
set.seed(0)
my.counts <- DropletUtils:::simCounts()
set.seed(0)
out <- emptyDrops(my.counts, lower=lower)
set.seed(0)
out.ambient <- emptyDrops(my.counts,test.ambient=T, lower=lower)
sum(out$FDR<fdr & out$Total>lower)
# 901
sum(out.ambient$FDR<fdr & out.ambient$Total>lower)
# 801
The documentation for test.ambient says whether results for barcodes with totals less or equal to lower should be reported, so the number of significant barcodes with totals above lower should be the same. Am I missing something?
Hi all,
I am trying to create a SingleCellExperiment object to use in analysis. I downloaded the following two types of files from NCBI-GEO, but I'm not sure how to use this to make a SingleCellExperiment object.
filtered_feature_bc_matrix.h5
filtered_contig_annotations.csv
I tried to use the following function
read10xCounts(file, col.names=FALSE, type=c("HDF5"), group=NULL)
But I got the following error
Error in h5read(path, paste0(group, "/genes")) : Object 'matrix/genes' does not exist in this HDF5 file.
Any thoughts would be greatly appreciated
I get the following error when trying to use the swappedDrops() function.
sPaths <- c("/Volumes/@home/scRNA/bin/cellRanger/pilot2/Pilot2Library1/outs/filtered_feature_bc_matrix.h5", "/Volumes/@home/scRNA/bin/cellRanger/pilot2/Pilot2Library2/outs/filtered_feature_bc_matrix.h5")
sD <- swappedDrops(sPaths)
HDF5-DIAG: Error detected in HDF5 (1.10.3) thread 0:
#000: H5D.c line 292 in H5Dopen2(): unable to open dataset
major: Dataset
minor: Can't open object
#1: H5Dint.c line 1205 in H5D__open_name(): not found
major: Dataset
minor: Object not found
#2: H5Gloc.c line 422 in H5G_loc_find(): can't find object
major: Symbol table
minor: Object not found
#3: H5Gtraverse.c line 851 in H5G_traverse(): internal path traversal failed
major: Symbol table
minor: Object not found
#4: H5Gtraverse.c line 627 in H5G__traverse_real(): traversal operator failed
major: Symbol table
minor: Callback failed
#5: H5Gloc.c line 378 in H5G__loc_find_cb(): object 'barcode' doesn't exist
major: Symbol table
minor: Object not found
Error in read10xMolInfo(samples[i], barcode.length = barcode.length) :
c++ exception (unknown reason)
I've tried it on both the 'filtered_feature_bc_matrix.h5' and 'molecule_info.h5' files generated with cellRanger count (v 3.0.2) and get the same error. When I do the simulated reads as shown in the example everything works as expected.
Hi,
Is there any chance swappedDrops could be run on STARSolo output?
Currently STARSolo outputs barcodes.tsv, features.tsv and matrix.mtx but no h5 files.
While swappedDrops requires h5 file
Thanks
The error message
Warning: The following arguments are not used: drop
Error in CellsByIdentities(object = object, cells = cells) :
Cannot find cells provided
pbmc5k dataset, I have tried both the raw and filtered datasets.
Preceding code
pbmc.data <- Read10X(data.dir = "data/pbmc5k/raw_feature_bc_matrix/")
pbmc_raw <- CreateSeuratObject(counts = pbmc.data, project = "pbmc5k", min.cells = 3, min.features = 200)
pbmc_raw[["percent.mt"]] <- PercentageFeatureSet(pbmc_raw, pattern = "^MT-")
I have also tried without min.cells = 3, min.features = 20, to no avail.
Calling EmptyDrops
emptyDrops(
pbmc_raw,
lower = 100,
niters = 10000,
test.ambient = FALSE,
ignore = NULL,
alpha = NULL,
BPPARAM = SerialParam()
)
Similarly, I have tried testEmptyDrops, only to receive the same error.
RStudio Version 1.3.1056
Seurat_3.2.0
DropletUtils_1.6.1
Does anyone know why it's not working, and/or how I could get around the error?
the description says: "which shows the (log-)total UMI count for each barcode on the x-axis and the (log-)rank on the y-axis" and the figure shows the opposite, doesn't-it? (section 4 in the vignette)
Benefits from being able to operate with fewer HTOs, see https://support.bioconductor.org/p/9134854/#9134859. Probably gets around the first assumption mentioned there. However, library size variation blurs the distinguishing lines between doublets, confident singlets and non-confident singlets, so you may see more errors there.
Hi Aaron,
This is the usual appearance of the barcodeRanks plot of my single-cell datasets when I apply the default features of barcodeRanks:

However there is a precise batch in which the inflection point includes many more cells than before, going up to the second "knee" that my data has:

Would it be posible to restrict the inflection point to the first knee as before without changing the knee point? I've modified barcodeRanks parameters such as df and fit.bounds, however I only managed to change the spline or move the knee point to the second knee rather than restrict the inflection point.
Thank you for your help.
Best,
Lucia
Hi Aaron,
I used read10xCounts(..., type = "HDF5") to load a HDF5 file from CellRanger v3.
I was surprised to get an error when I then ran scater::calculateQCMetrics() on the result.
Ultimately, I think the issue is that read10xCounts(..., type = "HDF5") doesn't return the counts matrix as a 'pristine' TENxMatrix.
I encountered this using v1.4.1 of DropletUtils and can't test on devel right now, but I didn't see any recent changes around this behaviour.
This example hopefully illustrates the issue.
What do you think?
Thanks!
suppressPackageStartupMessages(library(DropletUtils))
example("write10xCounts", echo = FALSE)
#>
#> Attaching package: 'Matrix'
#> The following object is masked from 'package:S4Vectors':
#>
#> expand
tmpfile <- tempfile(fileext = ".h5")
write10xCounts(tmpfile, my.counts, gene.id=gene.ids,
gene.symbol=gene.symb, barcodes=cell.ids,
type="HDF5", version="3")
sce <- read10xCounts(tmpfile)
# This error was the first indication something wasn't as I expected.
scater::calculateQCMetrics(sce)
#> Registered S3 methods overwritten by 'ggplot2':
#> method from
#> [.quosures rlang
#> c.quosures rlang
#> print.quosures rlang
#> Error in (function (exprs_mat, start, end, all_feature_sets, all_cell_sets, : Evaluation error: netSubsetAndAperm() is not supported on a DelayedArray object with
#> multiple seeds at the moment. Note that you can check the number
#> of seeds with nseed()..
# I expected this to be a TENxMatrix.
class(counts(sce, withDimnames = FALSE))
#> [1] "DelayedMatrix"
#> attr(,"package")
#> [1] "DelayedArray"
# It almost is, but what's with the delayed ops in here?
str(counts(sce, withDimnames = FALSE))
#> Formal class 'DelayedMatrix' [package "DelayedArray"] with 1 slot
#> ..@ seed:Formal class 'DelayedAbind' [package "DelayedArray"] with 2 slots
#> .. .. ..@ along: int 2
#> .. .. ..@ seeds:List of 1
#> .. .. .. ..$ :Formal class 'DelayedDimnames' [package "DelayedArray"] with 2 slots
#> .. .. .. .. .. ..@ dimnames:List of 2
#> .. .. .. .. .. .. ..$ : int -1
#> .. .. .. .. .. .. ..$ : NULL
#> .. .. .. .. .. ..@ seed :Formal class 'TENxMatrixSeed' [package "HDF5Array"] with 5 slots
#> .. .. .. .. .. .. .. ..@ filepath : chr "/tmp/RtmpDw8AyK/file28625fac52e3.h5"
#> .. .. .. .. .. .. .. ..@ group : chr "matrix"
#> .. .. .. .. .. .. .. ..@ dim : int [1:2] 100 10
#> .. .. .. .. .. .. .. ..@ dimnames :List of 2
#> .. .. .. .. .. .. .. .. ..$ : NULL
#> .. .. .. .. .. .. .. .. ..$ : chr [1:10] "BARCODE-1" "BARCODE-2" "BARCODE-3" "BARCODE-4" ...
#> .. .. .. .. .. .. .. ..@ col_ranges:'data.frame': 10 obs. of 2 variables:
#> .. .. .. .. .. .. .. .. ..$ start: num [1:10] 1 101 200 298 397 496 595 693 791 891
#> .. .. .. .. .. .. .. .. ..$ width: int [1:10] 100 99 98 99 99 99 98 98 100 100
# Forcing the assay to be a TENxMatrix means scater::calculateQCMetrics() works.
counts(sce, withDimnames = FALSE) <- as(my.counts, "TENxMatrix")
scater::calculateQCMetrics(sce)
#> class: SingleCellExperiment
#> dim: 100 10
#> metadata(0):
#> assays(1): counts
#> rownames(100): ENSG00001 ENSG00002 ... ENSG000099 ENSG0000100
#> rowData names(10): ID Symbol ... total_counts log10_total_counts
#> colnames: NULL
#> colData names(11): Sample Barcode ...
#> pct_counts_in_top_200_features pct_counts_in_top_500_features
#> reducedDimNames(0):
#> spikeNames(0):Created on 2019-06-15 by the reprex package (v0.3.0)
devtools::session_info()
#> ─ Session info ──────────────────────────────────────────────────────────
#> setting value
#> version R version 3.6.0 (2019-04-26)
#> os Ubuntu 18.04.2 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en_AU:en
#> collate en_AU.UTF-8
#> ctype en_AU.UTF-8
#> tz Australia/Melbourne
#> date 2019-06-15
#>
#> ─ Packages ──────────────────────────────────────────────────────────────
#> package * version date lib source
#> assertthat 0.2.1 2019-03-21 [3] CRAN (R 3.5.3)
#> backports 1.1.4 2019-04-10 [3] CRAN (R 3.5.3)
#> beachmat 2.0.0 2019-05-02 [1] Bioconductor
#> beeswarm 0.2.3 2016-04-25 [1] CRAN (R 3.6.0)
#> Biobase * 2.44.0 2019-05-02 [1] Bioconductor
#> BiocGenerics * 0.30.0 2019-05-02 [1] Bioconductor
#> BiocNeighbors 1.2.0 2019-05-02 [1] Bioconductor
#> BiocParallel * 1.18.0 2019-05-03 [1] Bioconductor
#> BiocSingular 1.0.0 2019-05-02 [1] Bioconductor
#> bitops 1.0-6 2013-08-17 [3] CRAN (R 3.5.0)
#> callr 3.2.0 2019-03-15 [3] CRAN (R 3.5.3)
#> cli 1.1.0 2019-03-19 [3] CRAN (R 3.5.3)
#> colorspace 1.4-1 2019-03-18 [3] CRAN (R 3.5.3)
#> crayon 1.3.4 2017-09-16 [3] CRAN (R 3.5.0)
#> DelayedArray * 0.10.0 2019-05-02 [1] Bioconductor
#> DelayedMatrixStats 1.6.0 2019-05-02 [1] Bioconductor
#> desc 1.2.0 2018-05-01 [3] CRAN (R 3.5.0)
#> devtools 2.0.2 2019-04-08 [1] CRAN (R 3.6.0)
#> digest 0.6.19 2019-05-20 [3] CRAN (R 3.6.0)
#> dplyr 0.8.1 2019-05-14 [3] CRAN (R 3.6.0)
#> dqrng 0.2.1 2019-05-17 [1] CRAN (R 3.6.0)
#> DropletUtils * 1.4.1 2019-05-31 [1] Bioconductor
#> edgeR 3.26.4 2019-05-27 [1] Bioconductor
#> evaluate 0.14 2019-05-28 [3] CRAN (R 3.6.0)
#> fs 1.3.1 2019-05-06 [3] CRAN (R 3.6.0)
#> GenomeInfoDb * 1.20.0 2019-05-02 [1] Bioconductor
#> GenomeInfoDbData 1.2.1 2019-05-07 [1] Bioconductor
#> GenomicRanges * 1.36.0 2019-05-02 [1] Bioconductor
#> ggbeeswarm 0.6.0 2017-08-07 [1] CRAN (R 3.6.0)
#> ggplot2 3.1.1 2019-04-07 [3] CRAN (R 3.5.3)
#> glue 1.3.1 2019-03-12 [3] CRAN (R 3.5.3)
#> gridExtra 2.3 2017-09-09 [1] CRAN (R 3.6.0)
#> gtable 0.3.0 2019-03-25 [3] CRAN (R 3.5.3)
#> HDF5Array 1.12.1 2019-05-07 [1] Bioconductor
#> highr 0.8 2019-03-20 [3] CRAN (R 3.5.3)
#> htmltools 0.3.6 2017-04-28 [3] CRAN (R 3.5.0)
#> IRanges * 2.18.1 2019-05-31 [1] Bioconductor
#> irlba 2.3.3 2019-02-05 [1] CRAN (R 3.6.0)
#> knitr 1.23 2019-05-18 [3] CRAN (R 3.6.0)
#> lattice 0.20-38 2018-11-04 [4] CRAN (R 3.5.1)
#> lazyeval 0.2.2 2019-03-15 [3] CRAN (R 3.5.3)
#> limma 3.40.2 2019-05-17 [1] Bioconductor
#> locfit 1.5-9.1 2013-04-20 [1] CRAN (R 3.6.0)
#> magrittr 1.5 2014-11-22 [3] CRAN (R 3.5.0)
#> Matrix * 1.2-17 2019-03-22 [4] CRAN (R 3.5.3)
#> matrixStats * 0.54.0 2018-07-23 [1] CRAN (R 3.6.0)
#> memoise 1.1.0 2017-04-21 [1] CRAN (R 3.6.0)
#> munsell 0.5.0 2018-06-12 [3] CRAN (R 3.5.0)
#> pillar 1.4.1 2019-05-28 [3] CRAN (R 3.6.0)
#> pkgbuild 1.0.3 2019-03-20 [3] CRAN (R 3.5.3)
#> pkgconfig 2.0.2 2018-08-16 [3] CRAN (R 3.5.1)
#> pkgload 1.0.2 2018-10-29 [3] CRAN (R 3.5.1)
#> plyr 1.8.4 2016-06-08 [3] CRAN (R 3.5.0)
#> prettyunits 1.0.2 2015-07-13 [3] CRAN (R 3.5.0)
#> processx 3.3.1 2019-05-08 [3] CRAN (R 3.6.0)
#> ps 1.3.0 2018-12-21 [3] CRAN (R 3.5.2)
#> purrr 0.3.2 2019-03-15 [3] CRAN (R 3.5.3)
#> R.methodsS3 1.7.1 2016-02-16 [1] CRAN (R 3.6.0)
#> R.oo 1.22.0 2018-04-22 [1] CRAN (R 3.6.0)
#> R.utils 2.9.0 2019-06-13 [1] CRAN (R 3.6.0)
#> R6 2.4.0 2019-02-14 [3] CRAN (R 3.5.2)
#> Rcpp 1.0.1 2019-03-17 [3] CRAN (R 3.5.3)
#> RCurl 1.95-4.12 2019-03-04 [3] CRAN (R 3.5.2)
#> remotes 2.0.4 2019-04-10 [1] CRAN (R 3.6.0)
#> rhdf5 2.28.0 2019-05-02 [1] Bioconductor
#> Rhdf5lib 1.6.0 2019-05-02 [1] Bioconductor
#> rlang 0.3.4 2019-04-07 [3] CRAN (R 3.5.3)
#> rmarkdown 1.13 2019-05-22 [3] CRAN (R 3.6.0)
#> rprojroot 1.3-2 2018-01-03 [3] CRAN (R 3.5.3)
#> rsvd 1.0.1 2019-06-02 [1] CRAN (R 3.6.0)
#> S4Vectors * 0.22.0 2019-05-02 [1] Bioconductor
#> scales 1.0.0 2018-08-09 [3] CRAN (R 3.5.1)
#> scater 1.12.2 2019-05-24 [1] Bioconductor
#> sessioninfo 1.1.1 2018-11-05 [1] CRAN (R 3.6.0)
#> SingleCellExperiment * 1.6.0 2019-05-02 [1] Bioconductor
#> stringi 1.4.3 2019-03-12 [3] CRAN (R 3.5.3)
#> stringr 1.4.0 2019-02-10 [3] CRAN (R 3.5.2)
#> SummarizedExperiment * 1.14.0 2019-05-02 [1] Bioconductor
#> testthat 2.1.1 2019-04-23 [1] CRAN (R 3.6.0)
#> tibble 2.1.2 2019-05-29 [3] CRAN (R 3.6.0)
#> tidyselect 0.2.5 2018-10-11 [3] CRAN (R 3.5.1)
#> usethis 1.5.0 2019-04-07 [1] CRAN (R 3.6.0)
#> vipor 0.4.5 2017-03-22 [1] CRAN (R 3.6.0)
#> viridis 0.5.1 2018-03-29 [1] CRAN (R 3.6.0)
#> viridisLite 0.3.0 2018-02-01 [3] CRAN (R 3.5.0)
#> withr 2.1.2 2018-03-15 [3] CRAN (R 3.5.0)
#> xfun 0.7 2019-05-14 [3] CRAN (R 3.6.0)
#> XVector 0.24.0 2019-05-02 [1] Bioconductor
#> yaml 2.2.0 2018-07-25 [3] CRAN (R 3.5.1)
#> zlibbioc 1.30.0 2019-05-02 [1] Bioconductor
#>
#> [1] /home/peter/R/x86_64-pc-linux-gnu-library/3.6
#> [2] /usr/local/lib/R/site-library
#> [3] /usr/lib/R/site-library
#> [4] /usr/lib/R/libraryHi!
Thank you for the package.
I am sorry to bother, but I was wondering if the dimension difference I find between the single cell experiment object and the Seurat one is normal. Otherwise I may do something wrong.
To clarify:
`
#Single Cell Experiment
dirs <- list.dirs("data_scRNAseq", recursive = FALSE)
names(dirs) <- basename(dirs)
sce <- read10xCounts(dirs)
#Check sce dimension
dim(sce)
[1] 33538 1503347
#Seurat Object
#Load counts
dirs <- list.dirs("data_scRNAseq", recursive = FALSE)
#Loop into the directories with the matrix.mtx, barcodes.tsv, genes.tsv datasets and create a Seurat Object
for (dir in dirs){
seurat_data <- Read10X(data.dir = dir)
seurat_obj <- CreateSeuratObject(counts = seurat_data, min.features = 200, project = dir)
assign(dir, seurat_obj)
}
#Check dimensions
dim(seurat_obj)
[1] 33538 5438`
Could someone explain the dimension's difference? I am sorry if it's a naive question
`> sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods
[9] base
other attached packages:
[1] Seurat_3.2.0 scds_1.4.0
[3] scater_1.16.2 readxl_1.3.1
[5] Matrix_1.2-18 LSD_4.1-0
[7] ggplot2_3.3.2 DropletUtils_1.8.0
[9] cowplot_1.1.0 SingleCellExperiment_1.12.0
[11] SummarizedExperiment_1.20.0 Biobase_2.50.0
[13] GenomicRanges_1.42.0 GenomeInfoDb_1.26.1
[15] IRanges_2.24.0 MatrixGenerics_1.2.0
[17] matrixStats_0.57.0 S4Vectors_0.28.0
[19] BiocGenerics_0.36.0
loaded via a namespace (and not attached):
[1] plyr_1.8.6 igraph_1.2.6 lazyeval_0.2.2
[4] splines_4.0.2 BiocParallel_1.22.0 listenv_0.8.0
[7] digest_0.6.27 htmltools_0.5.0 viridis_0.5.1
[10] fansi_0.4.1 magrittr_2.0.1 tensor_1.5
[13] cluster_2.1.0 ROCR_1.0-11 limma_3.44.3
[16] globals_0.13.1 R.utils_2.10.1 colorspace_2.0-0
[19] rappdirs_0.3.1 ggrepel_0.8.2 dplyr_1.0.2
[22] crayon_1.3.4 RCurl_1.98-1.2 jsonlite_1.7.1
[25] spatstat_1.64-1 spatstat.data_1.4-3 survival_3.2-7
[28] zoo_1.8-8 ape_5.4-1 glue_1.4.2
[31] polyclip_1.10-0 gtable_0.3.0 zlibbioc_1.36.0
[34] XVector_0.30.0 leiden_0.3.3 DelayedArray_0.16.0
[37] BiocSingular_1.4.0 Rhdf5lib_1.10.1 future.apply_1.6.0
[40] HDF5Array_1.16.1 abind_1.4-5 scales_1.1.1
[43] edgeR_3.30.3 miniUI_0.1.1.1 Rcpp_1.0.5
[46] viridisLite_0.3.0 xtable_1.8-4 reticulate_1.18
[49] dqrng_0.2.1 rsvd_1.0.3 htmlwidgets_1.5.2
[52] httr_1.4.2 RColorBrewer_1.1-2 ellipsis_0.3.1
[55] ica_1.0-2 pkgconfig_2.0.3 R.methodsS3_1.8.1
[58] uwot_0.1.8 deldir_0.1-29 locfit_1.5-9.4
[61] tidyselect_1.1.0 rlang_0.4.8 reshape2_1.4.4
[64] later_1.1.0.1 munsell_0.5.0 cellranger_1.1.0
[67] tools_4.0.2 xgboost_1.2.0.1 cli_2.2.0
[70] generics_0.1.0 ggridges_0.5.2 stringr_1.4.0
[73] fastmap_1.0.1 goftest_1.2-2 fitdistrplus_1.1-1
[76] purrr_0.3.4 RANN_2.6.1 pbapply_1.4-3
[79] future_1.19.1 nlme_3.1-149 mime_0.9
[82] R.oo_1.24.0 compiler_4.0.2 rstudioapi_0.13
[85] beeswarm_0.2.3 plotly_4.9.2.1 png_0.1-7
[88] spatstat.utils_1.17-0 tibble_3.0.4 stringi_1.5.3
[91] lattice_0.20-41 vctrs_0.3.5 pillar_1.4.7
[94] lifecycle_0.2.0 lmtest_0.9-38 RcppAnnoy_0.0.17
[97] BiocNeighbors_1.6.0 data.table_1.13.2 bitops_1.0-6
[100] irlba_2.3.3 httpuv_1.5.4 patchwork_1.1.0
[103] R6_2.5.0 promises_1.1.1 KernSmooth_2.23-17
[106] gridExtra_2.3 vipor_0.4.5 codetools_0.2-16
[109] MASS_7.3-53 assertthat_0.2.1 rhdf5_2.32.4
[112] withr_2.3.0 sctransform_0.2.1 GenomeInfoDbData_1.2.4
[115] mgcv_1.8-33 grid_4.0.2 rpart_4.1-15
[118] tidyr_1.1.2 DelayedMatrixStats_1.10.1 Rtsne_0.15
[121] pROC_1.16.2 shiny_1.5.0 ggbeeswarm_0.6.0 `
Hi,
I'm encountering the following error when trying to use the read10xCounts function.
> library(DropletUtils)
> fname <- "data/pbmc4k/"
> list.files(fname)
[1] "barcodes.tsv" "genes.tsv" "matrix.mtx"
> sce <- read10xCounts(fname, col.names=TRUE)
Error in `rownames<-`(`*tmp*`, value = c("ENSG00000243485", "ENSG00000237613", :
invalid rownames length
I stepped through the function by hand and the error seems to occur in the final line, when constructing the SingleCellExperiment object. I had trouble finding the line that might be causing it in the SCE codebase; is this an error you've seen before, or do you know what might be causing it?
I'm using R 3.5.1, DropletUtils 1.2.1, and SingleCellExperiment 1.4.0. I'm observing this error in several of the publicly available 10X datasets, including the 10X PBMC 4k dataset that was used in the EmptyDrops paper.
Thanks!
Matt
I am trying to install this package in the Docker image from bioconductor/devel_core2:20190919 using BiocManager::install("DropletUtils"). But this installation failed.
The installation message of this package and the sessioninfo outputs are here: https://gist.github.com/kevinwang09/d65d57cf9d2c4ed8238bb648eb570f94
Any suggestions?
Thanks, Kevin
Hello,
First off, thank you so much for providing rigorous documentation for this R package; I've found this package to be incredibly useful for handling 10x datasets.
At your earliest convenience, I would greatly appreciate your advice on a specific DropletUtils function. Currently, I'm implementing the function 'downsampleReads' to obtain a downsampled UMI matrix that will allow me to properly benchmark my sample, processed with 10x V3 chemistry, against the same sample processed with Drop-Seq S^3 (second strand synthesis, updated protocol). While your function provides a downsampled UMI matrix, I'm wondering if there's a way to also output a downsampled Reads matrix? This way, I could doublecheck that my 10x downsampled dataset is at the same read depth as the Drop-Seq S^3 dataset I'm benchmarking against.
Thank you for your time.
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.