Metabuli is metagenomic classifier that jointly analyze both DNA and amino acid (AA) sequences. DNA-based classifiers can make specific classifications, exploiting point mutations to distinguish close taxa. AA-based classifiers have higher sensitivity in detecting homology between query and reference sequences, leverageing higher conservation of AA sequences. Metabuli combines the information of both sequence types using a novel k-mer structure, metamer, to enable both specific and sensitive characterization of metagenomic samples. In addition, it can classify reads against a database of any size as long as it fits in the hard disk.
For more details of Metabuli, please see Nature Methods, PDF, bioRxiv, or ISMB 2023 talk.

- Fixed a minor reproducibility issue.
- Fixed a performance-harming bug occurring with sequences containing lowercased bases.
- Auto adjustment of
--match-per-kmerparameter. Issue #20 solved. - Record version info. in
db.parameter
# install via conda
conda install -c conda-forge -c bioconda metabuli
# Linux AVX2 build (fast, recommended for most Linux system
# check using: cat /proc/cpuinfo | grep avx2)
wget https://mmseqs.com/metabuli/metabuli-linux-avx2.tar.gz; tar xvzf metabuli-linux-avx2.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
# Linux SSE2 build (slower, for old systems)
wget https://mmseqs.com/metabuli/metabuli-linux-sse2.tar.gz; tar xvzf metabuli-linux-sse2.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
# MacOS (Universal, works on Apple Silicon and Intel Macs)
wget https://mmseqs.com/metabuli/metabuli-osx-universal.tar.gz; tar xvzf metabuli-osx-universal.tar.gz; export PATH=$(pwd)/metabuli/bin/:$PATH
Metabuli also works on Linux ARM64 systems. Please check https://mmseqs.com/metabuli for static builds for other architectures.
To compile Metabuli from source code use the following commands:
git clone https://github.com/steineggerlab/Metabuli.git
cd Metabuli
mkdir build && cd build
cmake -DCMAKE_BUILD_TYPE=Release ..
make -j 16
The built binary can be found in ./build/src.
You can download pre-built databases using databases workflow.
NOTE: The databases workflow may not work if you don't use the latest version of Metabuli.
In that case, please manually download databases from this link.
Usage:
metabuli databases DB_NAME OUTDIR tmp
# NOTE
- A human genome (T2T-CHM13v2.0) is included in all databases except RefSeq_release.
- A human genome (GRCh38.p14) is included in RefSeq_release.
1. RefSeq Virus (8.1 GiB)
- NCBI RefSeq release 223 virus genomes
- Database will be in OUT_DIR/refseq_virus
metabuli databases RefSeq_virus OUT_DIR tmp
2. RefSeq Prokaryote and Virus (115.6 GiB)
- RefSeq prokaryote genomes (Complete Genome/Chromosome, 2024-03-26) + RefSeq Virus above.
- Database will be in OUT_DIR/refseq_prokaryote_virus
metabuli databases RefSeq OUTDIR tmp
3. GTDB (101 GiB)
- GTDB 214.1 (Complete Genome/Chromosome, CheckM completeness > 90 and contamination < 5).
- Database will be in OUT_DIR/gtdb
metabuli databases GTDB OUTDIR tmp
4. RefSeq Releases 217 (480.5 GiB) (OLD)
- Viral and prokaryotic genomes of RefSeq release 217 and human genome (GRCh38.p14)
metabuli databases RefSeq_release OUTDIR tmp
Downloaded files are stored in OUTDIR/DB_NAME directory, which can be provided for classify module as DBDIR.
metabuli classify <i:FASTA> <i:DBDIR> <o:OUTDIR> <Job ID> [options]
- INPUT : FASTA or FASTQ file of reads you want to classify.
- DBDIR : The directory of reference DB.
- OUTDIR : The directory where the result files will be generated.
- Job ID: It will be the prefix of result files.
# Paired-end
metabuli classify read_1.fna read_2.fna dbdir outdir jobid
# Single-end
metabuli classify --seq-mode 1 read.fna dbdir outdir jobid
# Long-read
metabuli classify --seq-mode 3 read.fna dbdir outdir jobid
* Important parameters:
--threads : The number of threads used (all by default)
--max-ram : The maximum RAM usage. (128 GiB by default)
--min-score : The minimum score to be classified
--min-sp-score : The minimum score to be classified at or below species rank.
--taxonomy-path: Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--reduced-aa : 0. Use 20 alphabets or 1. Use 15 alphabets to encode amino acids.
Give the same value used for DB creation.
--accession-level : Set 1 to use accession level classification (0 by default).
It is available when the DB is also built with accession level taxonomy.
- Paratemers for precision mode (Metabuli-P)
- Illumina short reads:
--min-score 0.15 --min-sp-score 0.5 - PacBio HiFi reads:
--min-score 0.07 --min-sp-score 0.3 - PacBio Sequel II reads:
--min-score 0.005 - ONT reads:
--min-score 0.008
- Illumina short reads:
This will generate two result files: JobID_classifications.tsv, JobID_report.tsv, and JobID_krona.html.
- Classified or not
- Read ID
- Taxonomy identifier
- Effective read length
- DNA level identity score
- Classification Rank
- List of "taxID : k-mer match count"
#Example
1 read_1 2688 294 0.627551 subspecies 2688:65
1 read_2 2688 294 0.816327 subspecies 2688:78
0 read_3 0 294 0 no rank
The proportion of reads that are assigned to each taxon.
#Example
33.73 77571 77571 0 no rank unclassified
66.27 152429 132 1 no rank root
64.05 147319 2021 8034 superkingdom d__Bacteria
22.22 51102 3 22784 phylum p__Firmicutes
22.07 50752 361 22785 class c__Bacilli
17.12 39382 57 123658 order o__Bacillales
15.81 36359 3 126766 family f__Bacillaceae
15.79 36312 26613 126767 genus g__Bacillus
2.47 5677 4115 170517 species s__Bacillus amyloliquefaciens
0.38 883 883 170531 subspecies RS_GCF_001705195.1
0.16 360 360 170523 subspecies RS_GCF_003868675.1
0.11 248 248 170525 subspecies RS_GCF_002209305.1
0.02 42 42 170529 subspecies RS_GCF_002173635.1
0.01 24 24 170539 subspecies RS_GCF_000204275.1
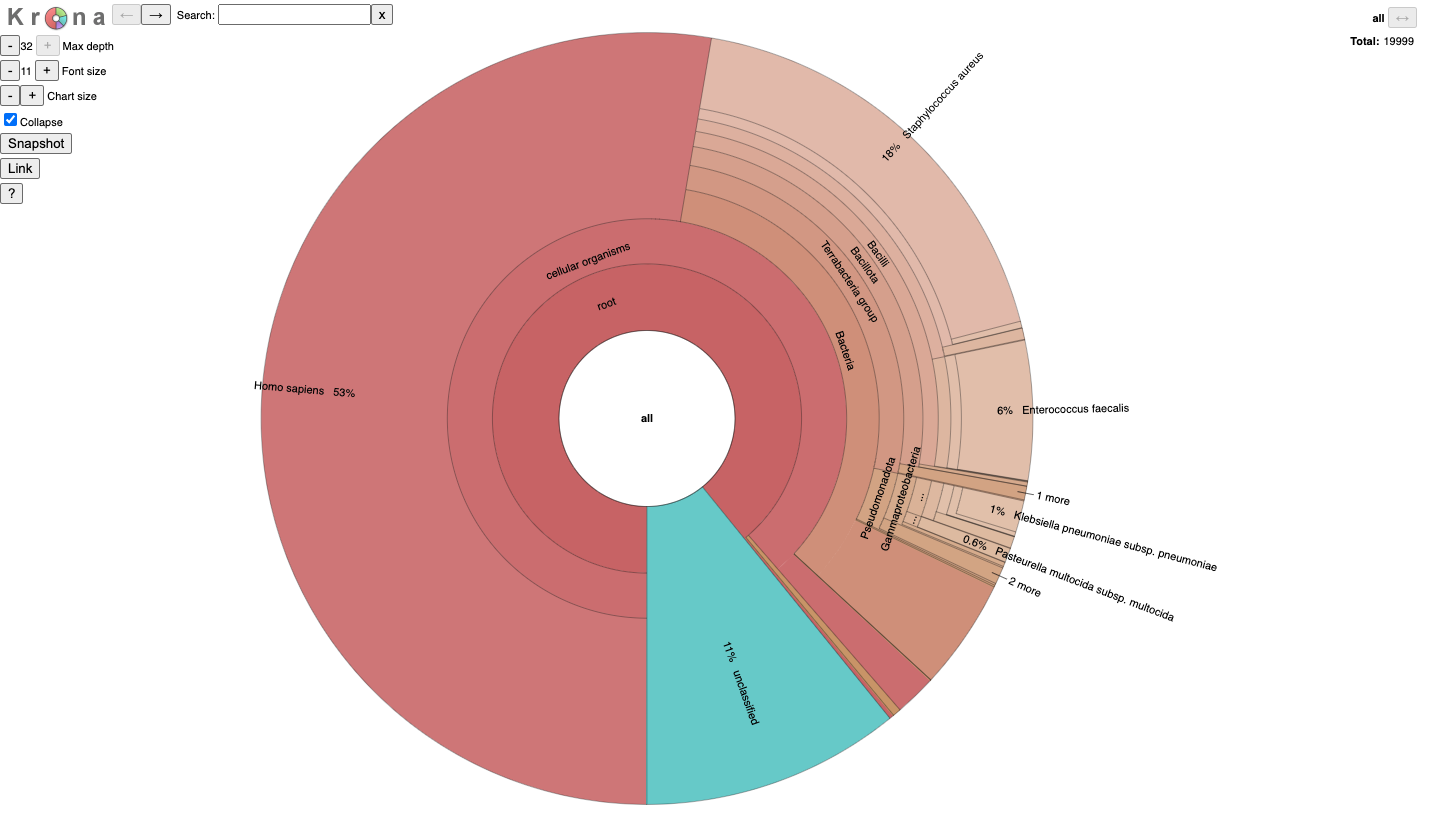
It is for an interactive taxonomy report (Krona). You can use any modern web browser to open JobID_krona.html.
Metabuli can classify reads against a database of any size as long as the database is fits in the hard disk, regardless of the machine's RAM size. We tested it with a MacBook Air (2020, M1, 8 GiB), where we classified about 1.5 M paired-end 150 bp reads (~5 GiB in size) against a database built with ~23K prokaryotic genomes (~69 GiB in size)
To build a custom database, you need three things:
- FASTA files : Each sequence of your FASTA files must be separated by '>accession.version' like '>CP001849.1'. The accession doesn't have to follow the NCBI format, but it must be unique and included in the accession2taxid file.
- accession2taxid : Mapping from accession to taxonomy ID. The sequences whose accessions are not listed here will be skipped.
- NCBI-style taxonomy dump : 'names.dmp' , 'nodes.dmp', and 'merged.dmp' are required. The sequences whose taxonomy IDs are not included here will be skipped.
The steps for building a database with NCBI or GTDB taxonomy are described below.
- If a custom sequence is to be included, edit
accession2taxidandtaxdumpfiles properly as follows.accession2taxid- For a sequence whose header is
>custom, addcustom[tab]custom[tab]taxid[tab]anynumber. - As above, version number is not necessary.
taxidmust be included in thenodes.dmpandnames.dmp.- Put any number for the last column. It is not used in Metabuli.
- For a sequence whose header is
taxdump- Edit
nodes.dmpandnames.dmpif you introduced a newtaxidinaccession2taxid.
- Edit
metabuli add-to-library <FASTA list> <accession2taxid> <DBDIR>
- FASTA list: A file containing absolute paths of each FASTA file.
- accession2taxid: A path to NCBI-style accession2taxid.
- DBDIR: Sequences will be stored in 'DBDIR/library'.
* Option
--taxonomy-path: Directory of taxdump files. (DBDIR/taxonomy by default)
* NOTE: When resume is needed, remove the files in DBDIR/library and run the command again.
It groups your sequences into separate files according to their species.
Accessions that are not included in the <accession2taxid> will be skipped and listed in unmapped.txt.
# Get the list of absoulte paths of files in your library
find <DBDIR>/library -type f -name '*.fna' > library-files.txt
metabuli build <DBDIR> <LIB_FILES> <accession2taxid> [options]
- DBDIR: The same DBDIR from the previous step.
- LIB_FILES: A file containing absolute paths of the FASTA files in DBDIR/library (library-files.txt)
- accession2taxid : A path to NCBI-style accession2taxid.
* Options
--threads : The number of CPU-cores used (all by default)
--taxonomy-path: Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--reduced-aa : 0. Use 20 alphabets or 1. Use 15 alphabets to encode amino acids.
--spacing-mask : Binary mask for spaced metamer. The same mask must be used for DB creation and classification. A mask should contain at least eight '1's, and '0' means skip.
--accession-level : Set 1 to use accession level taxonomy (0 by default).
This will generate diffIdx, info, split, and taxID_list and some other files. You can delete '*_diffIdx' and '*_info' if generated.
Requirements:
You need assembly FASTA files whose file name (or path) includes the assembly accession.
If you downloaded assemblies using ncbi-genome-download, you probably don't have to care about it.
The regular expression of assembly accessions is (GC[AF]_[0-9].[0-9])
# 1.
# 1-1. Move to the 'util' directory
cd METABULI_DIR/util
# 1-2. Run prepare_gtdb_taxonomy.sh
./prepare_gtdb_taxonomy.sh <DBDIR>
- DBDIR : Result files are stored in 'DBDIR/taxonomy'.
** Please clone Metabuli's repository to use this script.
** It is not provided in the precompiled binaries or bioconda package.
In DBDIR/taxonomy, it will generate taxonomy dmp files and assacc_to_taxid.tsv with other files.
# 2.
metabuli add-to-library <FASTA list> <accession2taxid> <DBDIR> --assembly true
- FASTA list : A file containing absolute paths of each assembly file.
Each path must include a corresponding assembly accession.
- accession2taxid : 'assacc_to_taxid.tsv' from the previous step
- DBDIR : The same DBDIR from the previous step.
** When resume is needed, remove the files in DBDIR/library and run the command again.
This will add your FASTA files to DBDIR/library according to their species taxonomy ID and generate 'my.accession2taxid'
# Get the list of absoulte paths of files in your library
find <DBDIR>/library -type f -name '*.fna' > library-files.txt
metabuli build <DBDIR> <LIB_FILES> <accession2taxid> [options]
- DBDIR: The same DBDIR from the previous step.
- <LIB_FILES>: A file containing absolute paths of the FASTA files in DBDIR/library (library-files.txt)
- accession2taxid : A path to NCBI-style accession2taxid.
* Options
--threads : The number of CPU-cores used (all by default)
--taxonomy-path: Directory where the taxonomy dump files are stored. (DBDIR/taxonomy by default)
--reduced-aa : 0. Use 20 alphabets or 1. Use 15 alphabets to encode amino acids.
--spacing-mask : Binary mask for spaced metamer. The same mask must be used for DB creation and classification.
A mask should contain at least eight '1's, and '0' means skip.
--accession-level : Set 1 to use accession level taxonomy (0 by default).
This will generate diffIdx, info, split, and taxID_list and some other files. You can delete '*_diffIdx' and '*_info' if generated.
Classifying RNA-seq reads from a COVID-19 patient to identify the culprit variant. The whole process must take less than 10 mins using a personal machine.
metabuli databases RefSeq_virus OUTDIR tmp
Option 1. Download using SRA Toolkit
fasterq-dump --split-files SRR14484345
Option 2. Download from web browser as FASTQ format
- link: https://trace.ncbi.nlm.nih.gov/Traces/?view=run_browser&page_size=10&acc=SRR14484345&display=download
- If the donwnloaded file includes both R1 and R2, use following commands.
cat SRR14484345.fastq | paste - - - - - - - - | tee >(cut -f 1-4 | tr "\t" "\n" > SRR14484345_1.fq) | cut -f 5-8 | tr "\t" "\n" > SRR14484345_2.fq
metabuli classify SRR14484345_1.fq SRR14484345_2.fq OUTDIR/refseq_virus RESULT_DIR JOB_ID --max-ram RAM_SIZE
Find a section like the example below
92.1796 510194 489403 no rank 2697049 Severe acute respiratory syndrome coronavirus 2
3.4290 18979 18979 subspecies 3000001 SARS-CoV-2 beta
0.2488 1377 1377 subspecies 3000003 SARS-CoV-2 gamma
0.0459 254 254 subspecies 3000000 SARS-CoV-2 alpha
0.0284 157 157 subspecies 3000004 SARS-CoV-2 omicron
0.0043 24 24 subspecies 3000002 SARS-CoV-2 delta