mikessh / mageri Goto Github PK
View Code? Open in Web Editor NEWMAGERI - Assemble, align and call variants for targeted genome re-sequencing with unique molecular identifiers
Home Page: http://mageri.readthedocs.org/en/latest/
License: Other
MAGERI - Assemble, align and call variants for targeted genome re-sequencing with unique molecular identifiers
Home Page: http://mageri.readthedocs.org/en/latest/
License: Other

























Hi,
I just started Mageri a few days ago to analyze some Illumina TruSeq Amplicon data from a custom panel with UMIs; however, I'm running into the following error a few minutes into the analysis:
Exception in thread "main" java.lang.OutOfMemoryError: Java heap space at java.util.concurrent.atomic.AtomicLongArray.<init>(AtomicLongArray.java:81) at com.antigenomics.mageri.core.mapping.QualitySumMatrix.<init>(QualitySumMatrix.java:35) at com.antigenomics.mageri.core.mapping.MutationsTable.<init>(MutationsTable.java:45) at com.antigenomics.mageri.core.mapping.ConsensusAligner.<init>(ConsensusAligner.java:60) at com.antigenomics.mageri.core.mapping.PConsensusAligner.<init>(PConsensusAligner.java:37) at com.antigenomics.mageri.core.mapping.PConsensusAlignerFactory.create(PConsensusAlignerFactory.java:34) at com.antigenomics.mageri.pipeline.analysis.PipelineConsensusAlignerFactory.create(PipelineConsensusAlignerFactory.java:53) at com.antigenomics.mageri.pipeline.analysis.ProjectAnalysis.run(ProjectAnalysis.java:107) at com.antigenomics.mageri.pipeline.Mageri.main(Mageri.java:129)
This occurs directly after building the UMI Index:
[Mon Aug 28 10:34:57 CEST 2017 +00m00s] [UMI_MAGERI_Test] Started analysis. [Mon Aug 28 10:34:57 CEST 2017 +00m00s] [UMI_MAGERI_Test] Pre-processing sample group MG357A. [Mon Aug 28 10:34:57 CEST 2017 +00m00s] [Indexer] Building UMI index, 0 reads processed, 0.0% extracted.. [Mon Aug 28 10:35:07 CEST 2017 +00m10s] [Indexer] Building UMI index, 588288 reads processed, 100.0% extracted.. [Mon Aug 28 10:35:17 CEST 2017 +00m20s] [Indexer] Building UMI index, 873247 reads processed, 100.0% extracted.. [Mon Aug 28 10:35:27 CEST 2017 +00m30s] [Indexer] Building UMI index, 1516289 reads processed, 100.0% extracted.. [Mon Aug 28 10:35:37 CEST 2017 +00m40s] [Indexer] Building UMI index, 2133652 reads processed, 100.0% extracted.. [Mon Aug 28 10:35:47 CEST 2017 +00m50s] [Indexer] Building UMI index, 2735621 reads processed, 100.0% extracted.. [Mon Aug 28 10:35:57 CEST 2017 +01m00s] [Indexer] Building UMI index, 3338909 reads processed, 100.0% extracted.. [Mon Aug 28 10:36:20 CEST 2017 +01m23s] [Indexer] Building UMI index, 3419451 reads processed, 100.0% extracted.. [Mon Aug 28 10:36:23 CEST 2017 +01m26s] [Indexer] Finished building UMI index, 3613540 reads processed, 100.0% extracted [Mon Aug 28 10:36:23 CEST 2017 +01m26s] [UMI_MAGERI_Test] Running analysis for sample group MG357A.
The command I'm running is:
java -Xmx100G -jar mageri.jar -R1 forward.fastq.gz -R2 reverse.fastq.gz --sample-name MG357 -O /path/to/output -M3 NNNNNN --references Homo_sapiens_assembly19.fasta --bed mybed.bed
I've tried to run with 32 GB first and then kept on going up until I reached the limit of my machine. I'm providing the Ensembl hg19 draft of the human genome as a fasta file as I want to avoid having to restrict my mapping to the panel targets.
Thank you for developing this useful tool! While running Mageri v1.1.1 on targeted DNA sequencing files using the M3 mode, I get the following error:
[Wed Nov 22 16:11:50 PST 2017 +00m00s] [MM082417-102117] Started analysis.
[Wed Nov 22 16:11:51 PST 2017 +00m00s] [MM082417-102117] Pre-processing sample group HIP21692-PBMC.
[Wed Nov 22 16:11:51 PST 2017 +00m00s] [Indexer] Building UMI index, 0 reads processed, 0.0% extracted..
[Wed Nov 22 16:12:01 PST 2017 +00m10s] [Indexer] Building UMI index, 199489 reads processed, 94.24% extracted..
[Wed Nov 22 16:12:11 PST 2017 +00m20s] [Indexer] Building UMI index, 387165 reads processed, 93.59% extracted..
[Wed Nov 22 16:12:21 PST 2017 +00m30s] [Indexer] Building UMI index, 586867 reads processed, 76.88% extracted..
[Wed Nov 22 16:12:31 PST 2017 +00m40s] [Indexer] Building UMI index, 785330 reads processed, 73.68% extracted..
[Wed Nov 22 16:12:42 PST 2017 +00m51s] [Indexer] Building UMI index, 971652 reads processed, 77.15% extracted..
[Wed Nov 22 16:12:52 PST 2017 +01m01s] [Indexer] Building UMI index, 1168617 reads processed, 79.61% extracted..
[Wed Nov 22 16:13:02 PST 2017 +01m11s] [Indexer] Building UMI index, 1366130 reads processed, 77.72% extracted..
[Wed Nov 22 16:13:12 PST 2017 +01m21s] [Indexer] Building UMI index, 1564702 reads processed, 79.74% extracted..
[Wed Nov 22 16:13:22 PST 2017 +01m31s] [Indexer] Building UMI index, 1766735 reads processed, 80.94% extracted..
[Wed Nov 22 16:13:32 PST 2017 +01m41s] [Indexer] Building UMI index, 1961040 reads processed, 75.08% extracted..
[Wed Nov 22 16:13:44 PST 2017 +01m53s] [Indexer] Building UMI index, 2131529 reads processed, 73.28% extracted..
[Wed Nov 22 16:13:54 PST 2017 +02m03s] [Indexer] Building UMI index, 2336328 reads processed, 75.06% extracted..
[Wed Nov 22 16:14:04 PST 2017 +02m13s] [Indexer] Building UMI index, 2537257 reads processed, 76.46% extracted..
[Wed Nov 22 16:14:14 PST 2017 +02m23s] [Indexer] Building UMI index, 2731940 reads processed, 77.45% extracted..
[Wed Nov 22 16:14:17 PST 2017 +02m26s] [Indexer] Finished building UMI index, 2797028 reads processed, 76.17% extracted
[Wed Nov 22 16:14:17 PST 2017 +02m26s] [MM082417-102117] Running analysis for sample group HIP21692-PBMC.
[Wed Nov 22 16:14:17 PST 2017 +02m26s] [MM082417-102117.HIP21692-PBMC] Assembling & aligning consensuses, 0 MIGs processed..
[Wed Nov 22 16:14:27 PST 2017 +02m36s] [MM082417-102117.HIP21692-PBMC] Assembling & aligning consensuses, 34560 MIGs processed..
[Wed Nov 22 16:14:37 PST 2017 +02m46s] [MM082417-102117.HIP21692-PBMC] Assembling & aligning consensuses, 85098 MIGs processed..
[Wed Nov 22 16:14:44 PST 2017 +02m53s] [MM082417-102117.HIP21692-PBMC] Finished, 95639 MIGs processed in total.
[Wed Nov 22 16:14:44 PST 2017 +02m53s] [MM082417-102117.HIP21692-PBMC] Calling variants.
Exception in thread "main" java.lang.NoSuchMethodError: java.util.concurrent.ConcurrentHashMap.keySet()Ljava/util/concurrent/ConcurrentHashMap$KeySetView;
at com.antigenomics.mageri.core.mapping.MutationsTable.getMutations(MutationsTable.java:170)
at com.antigenomics.mageri.core.variant.VariantCaller.(VariantCaller.java:80)
at com.antigenomics.mageri.pipeline.analysis.SampleAnalysis.run(SampleAnalysis.java:152)
at com.antigenomics.mageri.pipeline.analysis.ProjectAnalysis.run(ProjectAnalysis.java:111)
at com.antigenomics.mageri.pipeline.Mageri.main(Mageri.java:129)
Do you have any suggestions as to what may be causing this error?
Thank you,
David
submultiplexed, limit 10000 -> inspect checkout.txt, use sequences from SAM in IGV to fix your primersbiomart -> fa/bed.. but better include some script*.variant.caller.txt files
Consensus assembly appears to be quite slow for 1000+ reads/MIG cases
Suggestion: re-implement consensus assembly in a MiGEC style with remapping
VariantLibrary to infer error frequenciesHey Mike,
I am getting the following exception when running MAGERI:
[Fri Aug 11 12:22:47 EDT 2017 +00m00s] [my_project] Started analysis.
[Fri Aug 11 12:22:47 EDT 2017 +00m00s] [my_project] Pre-processing sample group my_sample.
[Fri Aug 11 12:22:47 EDT 2017 +00m00s] [Indexer] Building UMI index, 0 reads processed, 0.0% extracted..
Exception in thread "main" java.lang.RuntimeException: Error while parsing quality
at com.milaboratory.core.sequencing.io.fastq.SFastqReader.parse(SFastqReader.java:258)
at com.milaboratory.core.sequencing.io.fastq.PFastqReader.take(PFastqReader.java:213)
at com.antigenomics.mageri.core.input.PMigReader$PairedReaderWrapper.take(PMigReader.java:182)
at com.antigenomics.mageri.core.input.PMigReader$PairedReaderWrapper.take(PMigReader.java:166)
at cc.redberry.pipe.blocks.O2ITransmitter.run(O2ITransmitter.java:66)
at java.lang.Thread.run(Thread.java:744)
Caused by: com.milaboratory.core.sequence.quality.WrongQualityStringException: [-1]
at com.milaboratory.core.sequence.quality.SequenceQualityPhred.parse(SequenceQualityPhred.java:130)
at com.milaboratory.core.sequencing.io.fastq.SFastqReader.parse(SFastqReader.java:256)
... 5 more
I am running this on paired end reads.
My Read 1 looks like this:
@NB501788:53:H3HYMBGX3:1:11101:14803:1046 1:N:0:TACAGGTC+NATGCTGG UMI:GATCAC:CCCFFD
CTCCTNAGATACTGTTATCGTGCAGCGCNNNNNNNNNNNNNNNNNNNNNNNNNNTTAAAGAAATATGCA
+
AAAAA#EEEEEEEEEEEEEEEEEAEEEE##########################AEEEEEEEEEEEEEE
@NB501788:53:H3HYMBGX3:1:11101:11212:1046 1:N:0:TACAGGTC+NATGCTGG UMI:TGGAAT:CCCFFD
GCCGTNATGCAGTAGCAGCGAGGCATTCNNNNNNNNNNNNNNNNNNNNNNNNNNGGCTACTTCTTATACT
+
AAA/A#EEE/EEEEEEEE/EEEEEEA/E##########################EEEEEEEAAEEEEEEE
And Read 2 looks similar but with 2:N:0:....
Any help in this matter will be greatly appreciated.
Thanks,
Vaishnavi
Hi Mikessh, Thanks for you great tools. I was wondering how should I design the Master and Slave adapter to be compatible with illumina Exome Capture platform. Do you have any suggestion?
Are you ligating the Master and Slave adapter and then on a second step adding the Illumina Barcode?
Thanks!
Hi,
I have a quick question about your workflow. As far as I understand this pipeline requires at least 5 reads with the same UMI to be included in the analysis. Is there a flag to lower this value?
Thanks for developing this tool!
By applying read quality compression
Not properly handling read-through cases for long reads coming from short segments (e.g. MiSEQ)
As suggested, using machine learning & weka
subj.
I've just started using mageri and love the idea of an end-to-end UMI variant analysis tool. However, I'm having trouble trying to get it work with a sample containing 70M paired reads. This is a cfDNA sample processed with the Rubicon TagSeq kit (page 25 for UMI info) and sequenced on an Illumina NovaSeq using PE150. The UMIs are the first 6 bases of R1 and the last 6 of R2 of each read.
I used the following command to run mageri:
time java -Xms54G -Xmx240G -XX:ParallelGCThreads=4 -jar mageri.jar -M3 NNNNNN:NNNNNN --references ucsc.hg19.fasta -R1 S026-R1.fastq.gz -R2 S026-R2.fastq.gz out/After running for about 20 hours, mageri looks to be about 52M/70M or 74% done indexing, but progress has slowed to a halt as in the last 2 hours it has only processed 8 reads:
[Mon Mar 26 23:50:33 PDT 2018 +467m46s] [Indexer] Building UMI index, 52658259 reads processed, 99.99% extracted..
[Mon Mar 26 23:54:43 PDT 2018 +471m56s] [Indexer] Building UMI index, 52658260 reads processed, 100.0% extracted..
[Tue Mar 27 00:07:03 PDT 2018 +484m16s] [Indexer] Building UMI index, 52658267 reads processed, 100.0% extracted..
[Tue Mar 27 00:35:31 PDT 2018 +512m44s] [Indexer] Building UMI index, 52658277 reads processed, 100.0% extracted..
[Tue Mar 27 00:51:27 PDT 2018 +528m40s] [Indexer] Building UMI index, 52658280 reads processed, 100.0% extracted..
[Tue Mar 27 01:24:00 PDT 2018 +561m13s] [Indexer] Building UMI index, 52658285 reads processed, 99.99% extracted..
[Tue Mar 27 02:25:20 PDT 2018 +622m33s] [Indexer] Building UMI index, 52658287 reads processed, 100.0% extracted..
[Tue Mar 27 04:52:50 PDT 2018 +770m03s] [Indexer] Building UMI index, 52658292 reads processed, 100.0% extracted..
[Tue Mar 27 05:44:30 PDT 2018 +821m43s] [Indexer] Building UMI index, 52658297 reads processed, 100.0% extracted..
[Tue Mar 27 08:06:46 PDT 2018 +963m59s] [Indexer] Building UMI index, 52658306 reads processed, 100.0% extracted..
[Tue Mar 27 09:36:28 PDT 2018 +1053m41s] [Indexer] Building UMI index, 52658315 reads processed, 99.99% extracted..
[Tue Mar 27 11:41:53 PDT 2018 +1179m06s] [Indexer] Building UMI index, 52658323 reads processed, 99.99% extracted..
Of the 240Gb of RAM given, its currently only using about 107Gb.
I'm not sure I'm interpreting the output correctly and if this is to be expected, so I thought I'd ask. Is this normal? About how long should a sample of this size take to run? Any help is greatly appreciated!
We must also store binary output so users will be able to use oncomigec API for post-analysis.
Dear developer:
I encountered the following problem
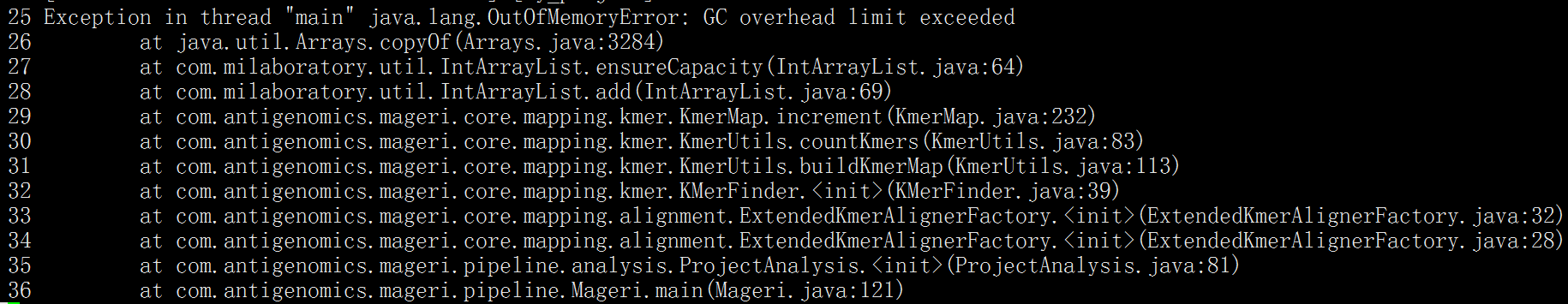
my fastq file like
@A00682:186:HLWT5DSXX:4:1101:3025:1000 1:N:0:CACCAGTT+CCAGTGTT
NCAATTTGATGTTGCAGTCTTGCAGGCCAATGATGGACACTCCTTCCATAATATACTGTTCCAAGTGAAGACCGTGCCTCAGGTAGGTGTCATTCCTAGAGTTACAGTTTCTTTGCACCTAGATGTGAACTTCTACGGTTGATATTATTA
+
#FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF::,FFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
my bed like
chr2 29416078 29416798
chr2 29419572 29419788
chr2 29420333 29420542
my hg19 is
>chr10
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
I don't know where the problem is, hope i can get help!Thanks!
I have run quiagen pannel and I have problems on detec the umi. No variant are detected.Even if I use --non-umi no result obtained.
I use the last release of mageri and I try to use M4 and select the primers from smCounter pipelines.
Could please help me?
Make sure that we have a unified output format, all variant coordinates and matrix numbering (headers) should be 1-based
for compatibility with gatk
Hi mikessh:
Thanks for sharing this great tool. I am trying to use mageri to call variants with the libraries containing UMIs. I have three fastq files generated from bcl2fastq. Two reads for pair ends, and one read for UMI.
This is how fastq file containing UMIs looks like:
@SN638:1063:HTVFJBCXX:1:1115:18632:43696 2:N:0:ATGCCTAA
GAGTTGCGCT
+
<<AG.GGIG<
This is the fastq for one of the pair end read:
@SN638:1063:HTVFJBCXX:1:1115:18632:43696 3:N:0:ATGCCTAA
GTGGAGTCACAGCGGAGATAGTGCCCTGGCCCTGGGCTTGTGGGGCTGCCCAGCAGCTGCCCATAAAGGACCTGATCGCTGGTGCCAGACTGGGATTGG
+
<GGGIIIIIIIIGIIIIIIIGIGGIIIIIIGIIIIIIIIIIIIIIGIIIIGIIIIIIGGGGGGGIIGGGIIIIGIIIIIIIIIIGGIIIIIIIIGAGGG
The read from pair end fastq does not contain UMI. Based on Mageri's manual, I am trying to use M4 option, but not sure how to modify the header. Any suggestion?
Thanks for your help,
Best
Rosemarie
Hi Mikhail,
I really enjoyed reading your Mageri paper, and it seems like a super useful tool. However, I have a practical question regarding the use of the master_adapter and slave_adapter. I would like to see a real example, so that I know more exactly what the adapters should look like. Do you only use a master_adapter with single end reads? Or when can you omit the slave_adapter? What does the master_first column mean exactly?
I have attached an example of one of my setups on which I would like to use your Mageri tool:
 . The starting point are TruSeq adapters where we added one UMI. Samples are pooled and will be sequenced using paired-end Illumina Next-Seq chemistry.
. The starting point are TruSeq adapters where we added one UMI. Samples are pooled and will be sequenced using paired-end Illumina Next-Seq chemistry.
Could you maybe point out some tips and tricks on how to set up the adapter sequences for Mageri?
Thanks a lot.
The variant calling Q score is capped at 100. Is there any options to relax or remove this cap? Currently it won't differentiate 0.1% and 1% variants -- both are given a QUAL = 100. Thank you!
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.