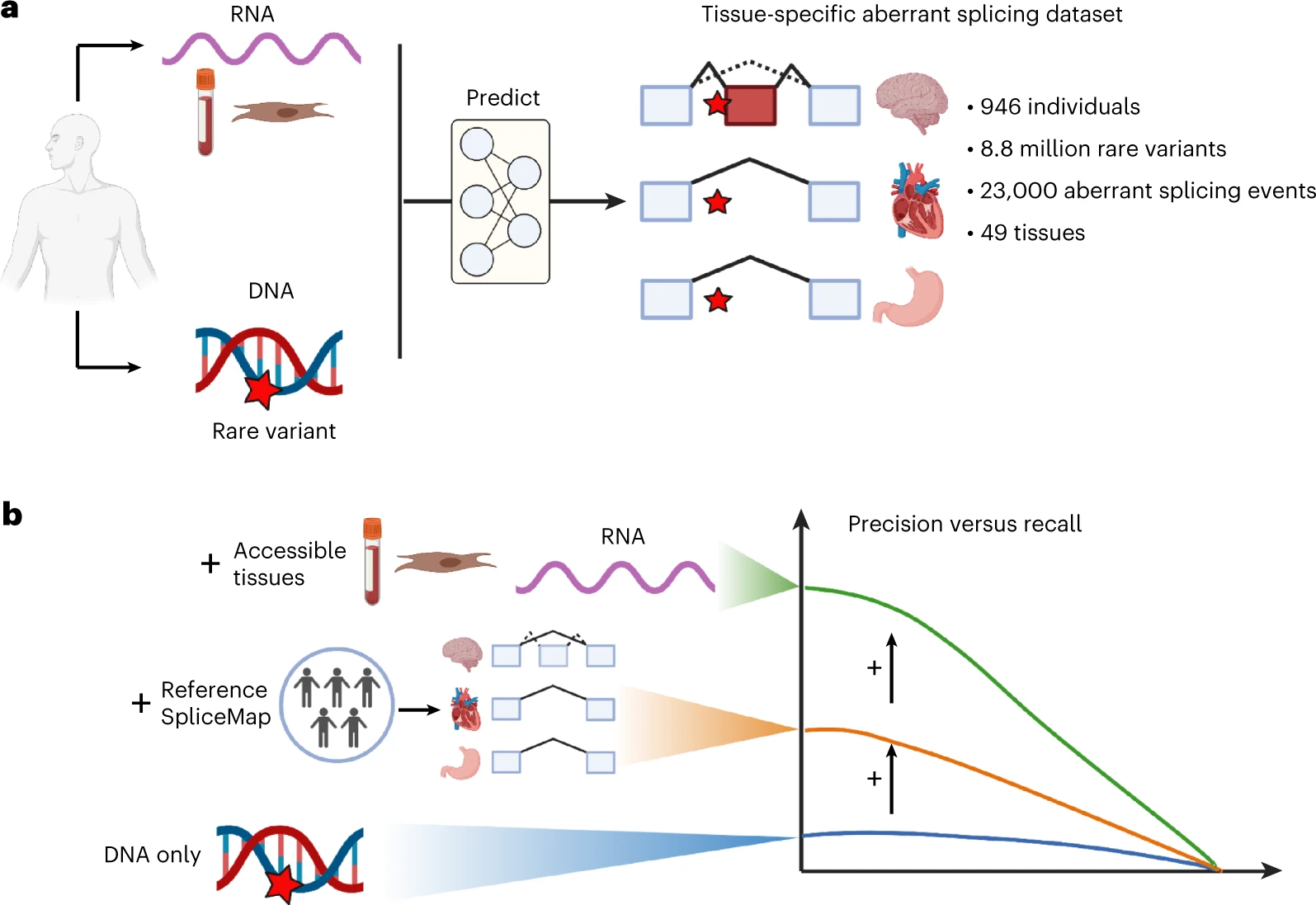
AbSplice is a method that predicts aberrant splicing across human tissues, as described in Wagner, Çelik et al., Nature Genetics 2023.
Precomputed AbSplice-DNA scores for all possible single-nucleotide variants genome-wide are available here (hg38) and here (hg19) for download.
AbSplice predictions are computed from VCF files and are based on enhanced tissue-specific splice site annotations (SpliceMaps). The scores represent the probability that a given variant causes aberrant splicing in a given tissue.
AbSplice-DNA: if only DNA is available, different DNA-based splicing predictions, as well as information from tissue-specific SpliceMaps, are combined into the integrative model AbSplice-DNA (see example use case).
AbSplice-RNA: if RNA-seq from clinically accessible tissues (e.g. blood or skin) is available, these direct splicing measurements can be used to predict aberrant splicing in another tissue from the same individual with the model AbSplice-RNA.
The source code to create and use SpliceMaps is under MIT license. Pre-computed SpliceMaps are under MIT license. The source code of AbSplice is under MIT license. The pre-trained AbSplice models as well as the pre-computed AbSplice scores are under the CC BY NC 4.0 license for academic and non-commercial use. This is because the implementation of running AbSplice predictions makes use of trained SpliceAI models which are currently under the CC BY NC 4.0 by Illumina (as of 21st December 2023).
Instead of Docker you can also use Podman (you just need to replace docker with podman in all the commands).
Download the image archive (file size is 5GB):
wget https://zenodo.org/record/8095625/files/absplice.oci
Load the image from archive:
docker load -i absplice.oci
Run the image with command line interface:
docker run -it --name absplice_container localhost/absplice:latest /bin/bash
Now you are working inside the container. The conda environment is already installed here, you just need to activate it:
conda activate absplice_dock
Clone the AbSplice repository to the container:
git clone https://github.com/gagneurlab/absplice.git
cd absplice
Clone git repo:
git clone https://github.com/gagneurlab/absplice.git
cd into repo directory:
cd absplice
Install conda environment:
# Recommended if you have mamba installed
mamba env create -f environment.yaml
# otherwise
conda env create -f environment.yaml
Activate conda environment:
conda activate absplice
Install modules from absplice:
pip install -e .
Note: if you run AbSplice on large datasets, you might experience memory issues with the full output. We suggest setting the config fields extra_info_dna and extra_info_rna to False in such cases and if you do not need additional information from SpliceAI and SpliceMaps (True by default). This will crop the output, leaving only the columns with AbSplice features and unique row identifiers.
The full output of AbSplice is tabular data with variant, gene_id, tissue being the unique row identifier. It contains the following columns:
| ID | Column | Description |
|---|---|---|
variant |
Variant | Variant as chrom:pos:ref>alt. |
gene_id |
GeneID | Ensembl GeneID of the gene which the variant belongs to. |
tissue |
Tissue | Name of the tissue that was used from SpliceMap. |
AbSplice_DNA |
AbSplice-DNA | The AbSplice score is a probability estimate of how likely aberrant splicing of some sort takes place in a given tissue and reports the splice site with the strongest effect. The model was trained using scores from MMSplice and SpliceAI models as well as annotation features from tissue-specific SpliceMaps. To ease downstream applications we suggest three cutoffs (high: 0.2, medium: 0.05, low: 0.01), which approximately have the same recalls as the high, medium and low cutoffs of SpliceAI. |
delta_score |
SpliceAI DeltaScore | Input feature to AbSplice. The main score predicted by SpliceAI, computed as a maximum of acceptor gain, acceptor loss, donor gain, and donor loss delta scores. The score represents probability of the variant being splice-altering. |
delta_logit_psi |
MMSplice + SpliceMap score | Input feature to AbSplice. The score is computed by using SpliceMap as an annotation for MMSplice. The score shows the effect of the variant on the inclusion level (PSI – percent spliced in) of the junction. The score is on a logit scale. If the score is positive, it shows that variant leads higher inclusion rate for the junction. If the score is negative, it shows that variant leads higher exclusion rate for the junction. |
delta_psi |
MMSplice + SpliceMap + Ψ_ref score | Input feature to AbSplice. The delta_psi (∆Ψ) score is computed by converting delta_logit_psi (∆𝑙𝑜𝑔𝑖𝑡(Ψ)) to natural scale with the splicing scaling law and ref_psi (Ψ𝑟𝑒𝑓): ∆Ψ = σ(∆𝑙𝑜𝑔𝑖𝑡(Ψ) + 𝑙𝑜𝑔𝑖𝑡(Ψ𝑟𝑒𝑓)) − Ψ𝑟𝑒𝑓 |
splice_site_is_expressed |
Splice site expressed | Input feature to AbSplice. Binary feature indicating if the splice site is expressed in the target tissue using a cutoff of 10 split reads (median_n from SpliceMap). |
junction |
Intron | Coordinates of the respective intron from SpliceMap. |
event_type |
Type of splicing event | The event_type takes values either psi3 or psi5 for alternative donor or alternative acceptor site usage (from SpliceMap). |
splice_site |
Splice site location | Coordinates of the splice site. |
ref_psi |
Ψ reference score | Reference level of site usage from SpliceMap. |
median_n |
Median coverage Splice site | The median number of split reads sharing the splice site (from SpliceMap). |
acceptor_gain |
SpliceAI Delta score (acceptor gain) | Probability computed by SpliceAI that the variant will lead to an acceptor gain at acceptor_gain_position . |
acceptor_loss |
SpliceAI Delta score (acceptor loss) | Probability computed by SpliceAI that the variant will lead to an acceptor loss at acceptor_loss_position. |
donor_gain |
SpliceAI Delta score (donor gain) | Probability computed by SpliceAI that the variant will lead to a donor gain at donor_gain_position. |
donor_loss |
SpliceAI Delta score (donor loss) | Probability computed by SpliceAI that the variant will lead to a donor loss at donor_loss_position. |
acceptor_gain_position |
SpliceAI Delta postion (acceptor gain) | Delta position represents the location of respective splicing change relative to the variant position: positive values are downstream of the variant, negative values are upstream. |
acceptor_loss_position |
SpliceAI Delta position (acceptor loss) | See description of acceptor_gain_position. |
donor_gain_position |
SpliceAI Delta postion (donor gain) | See description of acceptor_gain_position. |
donor_loss_position |
SpliceAI Delta position (donor loss) | See description of acceptor_gain_position. |
The example folder contains a snakemake workflow to generate AbSplice predictions, given a vcf file and a fasta file (either for hg19 or hg38, will be downloaded automatically).
The snakemake workflow will download precomputed SpliceMaps from Zenodo and run AbSplice based on these annotations.
To generate predictions run:
cd example/workflow
python -m snakemake -j 1 --use-conda
To run the workflow on your own data do the following:
-
Store all (or provide a symlink to) vcf files for analysis in
data/resources/analysis_files/vcf_files/. -
Specify the genome version that you are going to use (hg19 or hg38) in the field
genomeof the config file. -
In the field
splicemap_tissuesof the config file you can uncomment the tissues that AbSplice will use to generate predictions.
Optionally:
- If you want to run the example on large datasets, you can enable a fast lookup interface spliceai_rocksdb that uses precomputed SpliceAI scores.
The first time you use it, the precomputed database will be downloaded (it will take significant time – about 1 hour and use approximately 180GB of storage).
To enable fast lookup for SpliceAI simply change the field
use_rocksdbin the config file toTrue.
For users who work with the container:
To run AbSplice on your own vcf-files, you need to copy them from your disk to the container. If you are inside the container, run:
exit
To copy a vcf-file from your disk to the container run:
docker cp path/on/your/disk absplice_container:/app/absplice/example/data/resources/analysis_files/vcf_files/
To execute the container run:
docker start absplice_container
docker exec -it absplice_container /bin/bash
To edit the config file inside the container use pre-installed editor nano as follows (or optionally install any other editor):
nano config.yaml
To close the editor press Ctrl+X, choose whether to save the changes: Y or N, and press Enter.
AbSplice-RNA combines DNA and RNA information. For each individual, DNA-based predictions of variants will be combined with RNA-based predictions/ measurements for junctions in the vicinity of the variants. The input are vcf files (either single or multisample) from DNA and the results from running FRASER on RNA-seq samples using DROP.
The DNA IDs in the vcf file have to match the DNA IDs in the DNA_ID column of the sample annotation file from DROP.
Example output for AbSplice-RNA.
To run AbSplice-RNA on your own data you need to:
- Set the field
AbSplice_RNAof the config file toTrue. - Run the aberrant splicing module of DROP and copy the root directory of DROP into this folder.
- Specifiy the names of the DROP groups in the field
DROP_groupof the config file. - Speficy the gene annotation that was used to run DROP in the field
geneAnnotationof the config file.


































